
 PDF下载 ( 6007 KB)
PDF下载 ( 6007 KB)

HBV及其相关蛋白泛素化修饰研究进展
DOI: 10.3969/j.issn.1001-5256.2021.09.038
Research advances in hepatitis B virus and ubiquitination of related proteins
-
摘要: 蛋白质泛素化广泛存在于细胞内,是蛋白翻译后的一种修饰。HBV及其相关蛋白的泛素化研究越来越受到重视。对HBV及其相关蛋白的泛素化修饰进行了综述,为进一步了解HBV的复制调控及其蛋白的泛素化研究提供了借鉴,为治愈慢性HBV感染提供新的思路和方法。Abstract: Protein ubiquitination is widely observed in cells and is a modification after protein translation. Hepatitis B virus (HBV) and ubiquitination of related proteins have attracted more and more attention. This article reviews HBV and the ubiquitination of related proteins, so as to provide a reference for further research on the regulation of HBV replication and the ubiquitination of related proteins, as well as new ideas and methods for curing chronic HBV infection.
-
Key words:
- Hepatitis B Virus /
- Viral Proteins /
- Ubiquitination
-
HBV是目前已知感染人类最小的DNA病毒,大小为42 nm,基因组全长为3.2 kb,包含S、C、P、X 4个开放读码框架。病毒以共价闭合环状DNA(covalently closed circular DNA,cccDNA)为模板,采用宿主DNA依赖的RNA聚合酶,转录5种不同的mRNA,分别是3.5 kb的pgRNA、preC mRNA,2.4 kb、2.1 kb的preS/S mRNA和0.7 kb的HBx mRNA。pgRNA作为遗传物质包裹入核心颗粒,逆转录合成部分双链DNA基因组,并翻译病毒聚合酶蛋白(Pol);preC mRNA翻译出前C/C蛋白,进一步修饰后形成HBeAg、HBcAg;preS/S mRNA使用不同的起始密码子,翻译出3种包膜蛋白,分别是大蛋白(preS1+preS2+S)、中蛋白(preS2+S)和小蛋白(S);HBx mRNA翻译乙型肝炎病毒X蛋白(HBx)[1]。
蛋白质泛素化是蛋白翻译后的一种修饰,在细胞内广泛存在。细胞内80%~90%的蛋白质可以被泛素化系统识别和修饰[2]。蛋白质的泛素化由泛素蛋白酶体系统精密调控。泛素蛋白酶体系统由泛素、泛素活化酶(ubiquitin-activating enzyme, E1)、泛素结合酶(ubiquitin-conjugating enzyme, E2)、泛素连接酶(ubiquitin ligase, E3),以及26S蛋白酶体组成。泛素化过程大致为:E1首先以ATP依赖的方式与泛素结合,接下来泛素被转移到E2上;E3与蛋白底物相互作用,并将E2上的泛素转移至蛋白底物上,最终泛素化的底物进入26S蛋白酶体中进行降解[3]。E3在抗病毒天然免疫信号的各个环节发挥重要的调节作用,过程详见图 1[4]。
1. S基因编码蛋白
1.1 HBsAg相互作用蛋白
HBsAg是HBV包膜蛋白的主要成分,包括L(大蛋白)、M(中蛋白)、S(小蛋白)3种蛋白,HBV依赖L蛋白中小肽段与靶细胞的钠离子-牛磺酸共转运多肽(sodium taurocholate cotransporting polypeptide,NTCP)受体结合;L蛋白还具有病毒复制中的调节作用;M蛋白主要是装配病毒颗粒。外膜蛋白携带B淋巴细胞和T淋巴细胞表位,给宿主提供保护性免疫的免疫源。目前,关于HBsAg与细胞蛋白之间的相互作用还知之甚少,Toh等[5]利用酵母分裂泛素系统,分离了很多细胞膜蛋白,其中包括内质网的驻留蛋白(硫氧化还原蛋白相关跨膜蛋白2)、参与网格蛋白介导的内吞和HIV介导的CD4下调的衔接蛋白、柯萨奇B病毒的共同受体等,这些蛋白均与HBsAg存在相互作用。
Hartmann-Stühler等[6]利用L蛋白-特异性前S1结构域作为诱饵,启动了一个双杂交筛选。通过这种方法,证实了γ2衔接蛋白是网格蛋白衔接蛋白的成员之一,负责蛋白质的分类和运输,是L蛋白特异性的结合对象。亲和层析和免疫共沉淀证实了L蛋白和γ2衔接蛋白之间存在相互作用的证据,并且确认了结合位点位于L蛋白特异的前S1结构域和γ2衔接蛋白特异的耳部结构域。有趣的是,两者在转染细胞中的共同表达,导致L蛋白和γ2衔接蛋白在高尔基体上聚集,因此研究强烈支持两种蛋白存在生理上的关联。
1.2 HBsAg降解途径
为了探索HBV包膜蛋白的降解途径,有研究[7]观察了内质网质量控制系统对M蛋白的监测和分泌功能。结果发现野生型M蛋白以及分泌功能不全的M蛋白突变体可以从内质网有效地逆向转运,通过与泛素化无关的胞质蛋白酶体途径降解。并且,该泛素非依赖途径与抗原递呈无关,因为无论是野生型还是突变型M蛋白,都难以通过MHC-Ⅰ递呈。通过添加两个赖氨酸残基,可增加泛素化并导致抗原递呈增加。Liu等[8]以HBV M蛋白为模型,发现了一种新的、不依赖泛素化的蛋白酶体依赖性内质网相关蛋白降解途径。在HBV感染的过程中,病毒可能会利用该途径限制抗原递呈。
利用内质网葡萄糖苷酶的竞争性抑制剂,研究者发现在产生HBV包膜糖蛋白的组织培养中,L蛋白和M蛋白的糖基化和非糖基化形式的数量都大大减少;相反,S蛋白没有受到影响。L蛋白和M蛋白分泌的减少可能是由蛋白酶体降解途径介导的,而在具有功能性葡萄糖苷酶的细胞中没有检测到L蛋白和M蛋白的蛋白酶体降解[9]。
2. C基因编码蛋白
2.1 HBcAg相关泛素连接酶
HBcAg是病毒核衣壳的主要成分,是参与包装RNA、反转录、病毒装配和释放的多功能蛋白。HBV在感染细胞的出芽分泌过程中,可能使用细胞γ2-衔接蛋白和泛素连接酶Nedd4,通过与泛素结合以协调其组装和释放[10]。此前已有研究[6]发现γ2-衔接蛋白和HBV L蛋白之间存在相互作用。进一步研究还发现,同样的γ2-衔接蛋白和病毒核衣壳也存在相互作用,可以通过破坏HBV/γ2-衔接蛋白的相互作用,抑制病毒的产生。突变分析揭示:γ2-衔接蛋白的泛素结合活性存在特定的泛素作用基序,可以与核衣壳发生相互作用。HBcAg含有两个赖氨酸残基(K7和K96),K96位是一个潜在的泛素化靶点,对γ2-衔接蛋白的识别和病毒的产生是必不可少的。核衣壳通过其晚期结构域样PPAY序列,与核内Nedd4相互作用,提示了细胞泛素系统参与HBV组装。过表达无催化活性的Nedd4突变体可以降低HBV的释放,表明蛋白质泛素化在功能上参与了HBV的产生[11]。另一项研究[12]表明,在转染肝癌细胞系中,HBcAg泛素化主要发生在赖氨酸残基K7上,通过K29位泛素化进行翻译后修饰。
Np95/ICBP90样环指蛋白(Np95/ICBP90-like RING finger protein,NIRF)具有自身泛素化活性,是泛素连接酶的标志。泛素连接酶NIRF与HBcAg结合,导致蛋白酶体介导的HBcAg降解。NIRF下调HBcAg蛋白水平,导致HepG2.2.15细胞上清液中HBV颗粒减少。敲除NIRF,则显著增加内源性HBcAg蛋白水平,导致HBV释放[13]。研究结果表明,NIRF与HBcAg的相互作用,促进HBcAg在体内的降解。NIRF介导的泛素-蛋白酶体途径通过控制HBcAg水平,影响HBV颗粒的释放,提示NIRF可能参与HBV的成熟。还有研究探讨了NIRF对HBV复制的影响及其机制,证明NIRF抑制了转染pAAV-HBV1.3的HepG2细胞中HBV DNA的复制和HBsAg、HBeAg的分泌。在表达HBV的小鼠模型中,NIRF也抑制HBV的复制和分泌。NIRF还降低了HBV cccDNA结合的H3组蛋白的乙酰化[14],cccDNA的活性在体内和体外均受到组蛋白H3乙酰化状态改变的调控,进而影响其转录和HBV的复制。表明NIRF不仅通过与HBcAg的直接作用参与HBV的复制,而且通过降低与HBV cccDNA结合的H3组蛋白的乙酰化而调控HBV的复制。
2.2 HBcAg的泛素化与免疫增强
泛素是一种高度保守的小分子调控蛋白,其介导的抗原处理快速且高效,并刺激细胞介导的免疫反应。因此,泛素介导的抗原处理已广泛应用于慢性感染和癌症研究,以提高免疫应答。重组pUb-HBcAg DNA疫苗诱导免疫BALB/c小鼠产生特异性抗-HBc反应和特异性细胞毒性T淋巴细胞(CTL)反应,表明泛素可以作为一种分子佐剂来提高DNA疫苗的效力[15-16]。
慢性HBV感染的特点是辅助性T淋巴细胞(Th)1型细胞免疫和特异性T淋巴细胞应答的功能受损。转染了泛素-HBcAg的慢病毒能够有效诱导树突状细胞的成熟,成熟的树突状细胞有效诱导T淋巴细胞转化为Th1,并产生HBcAg特异性CTL,增加抗原特异性CD8+/IFNγ+T淋巴细胞的数量[17]。胞质转导肽(cytoplasmic transduction peptide,CTP)源自人类免疫缺陷病毒1转录蛋白反式激活体的蛋白转导域,能够有效地将生物分子传递到细胞质中。利用CTP的细胞穿透特性,泛素-HBcAg-CTP融合蛋白能够直接渗透到树突状细胞的细胞质中。融合蛋白不仅增加了树突状细胞表面分子的表达和T淋巴细胞的细胞因子分泌,而且诱导T淋巴细胞分化为特异性CTL,增强了抗病毒能力[18]。融合蛋白引起特异性CTL活性增强,降低了HBsAg和HBV DNA血清水平,降低了HBsAg和HBcAg在HBV转基因小鼠肝脏组织中的表达,提示具有治疗方面的价值[19]。泛素-HBcAg-CTP融合蛋白还能上调T淋巴细胞中Jak2、Tyk2、STAT1和STAT4的表达[20]。
2.3 HBeAg与泛素连接酶的相互作用
HBeAg是调节免疫的功能蛋白,也是病毒高水平复制的标志。HBeAg可干扰NF-κB活性,进而导致高病毒载量。Wang等[21]研究了HBeAg抑制IL-1β刺激的NF-κB活性,结果表明,HBeAg可以与NF-κB必须调节蛋白(NF-κB essential modulator,NEMO)作用。NEMO是一种与IκB激酶相关的调节亚基,调节NF-κB的激活。HBeAg抑制IL-1β诱导的肿瘤坏死因子受体相关因子6(tumor necrosis factor associated factor 6,TRAF6)依赖性K63相关泛素化的NEMO,从而下调NF-κB活性,促进病毒复制及持续性感染。研究者通过原代人肝细胞,HBV感染的HepG2-NTCP细胞和临床肝脏样本研究,进一步证明了HBeAg对NF-κB信号通路的抑制作用。
3. P基因编码蛋白
3.1 Pol蛋白相关泛素连接酶
Pol蛋白是一种决定病毒复制活性的聚合酶蛋白:末端蛋白是反转录负链DNA的引物;聚合酶激活pgRNA反转录DNA链;RNA酶H清除RNA-DNA杂交链中的RNA,合成正链DNA。在接受伊马替尼(imatinib)(一种Abl激酶抑制剂)或硼替佐米(bortezomib)(一种蛋白酶体抑制剂)治疗的患者中,经常有HBV再激活的报道,但这种重新激活的潜在机制尚不清楚。Hou等[22]报道Pol蛋白经Cdt2招募到Cullin-RING连接酶4(Cullin-RING ligase 4,CRL4),从而进行泛素化和蛋白酶体降解,这一过程是由c-Abl非受体酪氨酸激酶激发的。伊马替尼和硼替佐米可稳定病毒Pol蛋白,增加HBV感染细胞中的病毒载量,因此,这可以从机制上解释以上两种药物激活患者的HBV复制。
细胞凋亡抑制蛋白2(cellular inhibitor of apoptosis protein 2,cIAP2)属于凋亡抑制剂(IAP)家族,是一种有效的细胞死亡抑制剂,cIAP2具有羧基末端的RING指结构域,该结构域具有介导蛋白质泛素化的泛素连接酶活性。使用cDNA微阵列筛选肿瘤坏死因子诱导的细胞基因,具有强烈上调cIAP2抗HBV的活性[23]。进一步研究[24]证明,cIAP2可以显著降低HBV DNA复制中间体的水平,但不能降低总病毒RNA或核心蛋白的水平。Pol蛋白是cIAP2的靶标,过表达cIAP2可以降低Pol蛋白水平,而过表达cIAP2的E3连接酶缺陷型突变体(称为cIAP2*)则不能。进一步研究表明,cIAP2可以与Pol蛋白结合,促进其多聚泛素化,通过蛋白酶体途径进行降解。
三结构域(tripartite motif,TRIM)家族是一个新的E3连接酶家族,研究表明,某些TRIM蛋白具有抗HBV复制的活性,例如TRIM14,一种Ⅰ型干扰素(IFN)刺激基因,通过靶向HBx来控制HBV的复制;TRIM22,可抑制HBV核心启动子的活性而影响HBV复制;以及TRIM25,可通过IL-27抑制HBV复制。Mu等[25]发现,SPRY域负责TRIM21与HBV Pol蛋白之间的相互作用,Pol蛋白的TP域与TRIM21相互作用。研究者使用在线工具预测了潜在的Pol蛋白泛素化位点,通过对Pol蛋白间隔区中的K260和K283点突变,证实是Pol蛋白泛素化的赖氨酸位点。TRIM21介导了Pol蛋白K48位多聚泛素化,导致其通过泛素蛋白酶体途径降解。
3.2 Pol蛋白通过泛素化途径影响其他蛋白的稳定性
视黄酸诱导基因Ⅰ(retinoic acid inducible gene Ⅰ,RIG Ⅰ)介导产生的IFN能够被HBV抑制。HBV明显干扰IFNβ的产生和STING介导的抗病毒免疫,STING已被证实为外源DNA识别和抗病毒天然免疫的核心因素。筛选分析表明,HBV Pol蛋白能够抑制STING激活干扰素调节因子3(interferon regulatory factor 3,IRF3)和诱导IFNβ。在不改变STING表达水平的情况下,Pol蛋白被证明与STING存在物理联系,其逆转录酶区域可以显著降低STING的K63位多聚泛素化,在抑制IFNβ的产生方面发挥作用,这可能是HBV对抗天然免疫的一种新机制[26]。
4. X基因编码蛋白
4.1 HBx与蛋白酶体相互作用
HBx是一种多功能的调节因子,已知能激活转录、影响DNA修复、调节细胞的生长和死亡。HBx蛋白在病毒复制、肝细胞癌变和激活某些信号转导途径方面十分重要。研究表明HBx是蛋白酶体的抑制剂。通过免疫共沉淀和蔗糖梯度离心共定位研究,发现HBx与26S蛋白酶体复合物存在关联[27]。HBx在HepG2细胞中的表达导致蛋白酶体的糜蛋白酶和类胰蛋白酶活性降低,泛素化溶菌酶水解也减少。通过杂交和免疫沉淀法,检测到HBx与蛋白酶体的PSMC1(S4 ATP酶)和PSMA7(α1/C2)亚基存在相互作用,并在蔗糖梯度离心时与蛋白酶体共沉积。另有研究[28]证实染色质重构因子BAF155是HBx的绑定伙伴之一。BAF155通过与20S蛋白酶体亚基PSMA7竞争结合HBx,使HBx免受泛素非依赖性蛋白酶体降解。
4.2 HBx降解相关泛素连接酶
Zhang等[29]开发了一种针对HBx的细胞穿透性全分子抗体(9D11-Tat)。内化的9D11-Tat抗体可通过Fc结合受体TRIM21介导蛋白质降解,显著降低细胞内HBx,促进了宿主细胞的抗病毒状态。泛素连接酶Siah-1可以靶向HBx进行多泛素化和蛋白酶体降解,并减弱HBx的转录活性[30]。在HepG2细胞中,敲除p53基因可以下调Siah-1水平,随后可见HBx水平上调[31]。而在Hep3B细胞中,异常表达的p53基因上调了Siah-1水平,同时可见HBx水平下调。HBx通过一个涉及p53和Siah-1的负反馈回路调节自身的蛋白水平,以控制HBV的复制。MARCH5是一种线粒体泛素连接酶,其高表达与肝癌患者生存率的提高有关[32]。MARCH5与在线粒体中聚集的HBx蛋白相互作用,并将其靶向降解。在MARCH5高表达的情况下,HBx诱导的活性氧自由基、线粒体自噬和环氧合酶-2基因表达受到抑制。
4.3 HBx通过E3泛素连接酶影响其他蛋白质
在HBx的凋亡与泛素化研究[33]中,发现HBx能够增强肝细胞对肿瘤坏死因子相关凋亡诱导配体的敏感性,其敏感性的增加与HBx诱导miR-125a上调有关,而miR-125a又抑制了其靶基因A20泛素连接酶的表达。在肝细胞中拮抗miR-125a或外源性表达A20,可消除HBx的促凋亡作用。原癌基因垂体肿瘤转化基因1(pituitary tumor transforming gene1,PTTG1)在肝细胞癌中呈高表达。体外实验[34]表明,HBx诱导PTTG1蛋白显著积累。HBx与PTTG1和/或Skp1-Cul1-F-box泛素连接酶复合物(Skp1-Cul1-F-box protein,SCF)的蛋白-蛋白相互作用,导致PTTG1与SCF之间的作用被部分破坏,阻碍其经蛋白酶体的降解。HBx介导Myc的稳定表达在病毒致癌中起着重要作用。Myc癌蛋白的稳定性受SCF泛素连接酶的调控,特别是其中的SCFSkp2或SCFFbw7泛素连接酶。通过免疫共沉淀和免疫荧光实验,揭示了HBx与Myc蛋白存在相互作用,进一步分析表明,HBx通过阻断Skp2稳定Myc癌蛋白[35]。
4.4 HBx的多重功能
HBx通过与宿主细胞胞质和胞核中的多种蛋白质相互作用以执行多方面的功能。HBx与损伤特异性DNA损伤结合蛋白1(DNA damage-binding protein 1,DDB1)的结合对于HBx激活HBV转录是至关重要的。DDB1与CUL4形成泛素连接酶,并与DDB1 cullin相关因子的衔接子亚基结合,靶向泛素化底物,在DNA修复、复制和转录中发挥重要作用。HBx被称为HBV复制的中枢调节因子,通过H-box基序与CUL4-DDB1泛素连接酶相互作用[36]。HBx在不同的CUL4-DDB1复合物背景中,以正向和负向的方式调节DDB1的功能,在HBV复制周期中起着不同的作用。
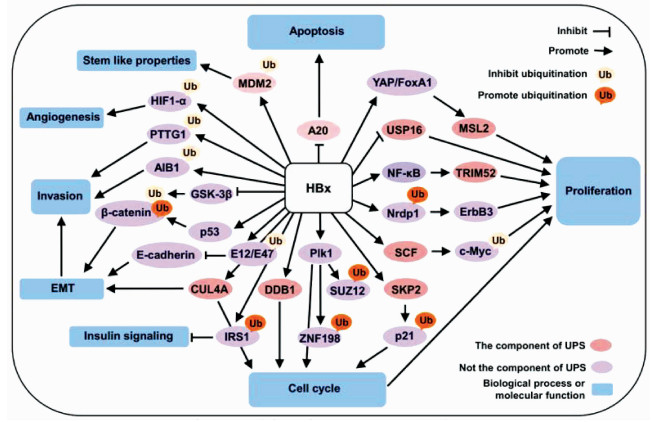
除TANK结合激酶1(TANK-binding kinase 1,TBK1)以外,HBx作为一种去泛素化酶能够从多种蛋白质中切割赖氨酸63连接的多泛素链,结合和解偶联RIG Ⅰ及TRAF3,导致它们从下游的衔接蛋白CARDIF或TBK1激酶上分离。除RIG Ⅰ和TRAF3外,HBx还与CARDIF、TRIF、NEMO、TBK1、B淋巴细胞kappa轻链多肽基因增强子抑制剂、epsilon激酶(IKKi)和IRF3相互作用。HBx可以靶向多个信号通路点,负调控Ⅰ型IFN的产生。MAVS是病毒激活信号通路的重要组成部分,激活NF-κB和IRF3诱导Ⅰ型IFN的产生[37]。HBx与MAVS相互作用,通过MAVS蛋白中的136位赖氨酸泛素化促进MAVS的降解,从而阻止诱导IFNβ[38]。HBx介导的发病机理中与泛素蛋白酶体系统有关的机制详见图 2[39]。
5. 与HBV cccDNA相关的泛素化研究
5.1 UBE2L3诱导降解载脂蛋白BmRNA编辑酶催化多肽3A(APOBEC3A)维持cccDNA的稳定
HBV cccDNA是编码所有HBV RNA的唯一模板,是导致病毒持续感染和复发的主要原因,清除HBV cccDNA是根治HBV持续感染的关键[40]。SNP rs59391722位于UBE2L3基因的启动子区域,该基因编码E2泛素结合酶,也称为UbcH7。全基因组关联研究证实,UBE2L3基因与成人慢性HBV感染的易感性增加有关。在儿童HBV感染者中,血清UBE2L3蛋白水平与HBV载量和HBeAg水平呈正相关。在HBV感染的HepG2-NTCP和人原代肝细胞中,UBE2L3基因敲低显著降低总HBV RNA、3.5 kb RNA以及cccDNA的水平。研究其机制发现,UBE2L3通过诱导蛋白酶体依赖性途径降解APOBEC3A以维持HBV cccDNA的稳定,而APOBEC3A主要负责HBV cccDNA的降解。此外,IFNα治疗后明显降低UBE2L3的表达,而沉默UBE2L3基因则增强了IFNα对HBV RNA、cccDNA和DNA的抗病毒活性[41]。
5.2 ITCH-NUMB通过负调控Notch信号通路抑制cccDNA
Notch信号通路活化后,通过两次蛋白水解切割(α/γ分泌酶)释放跨膜受体的细胞内结构域(Notch intracellular domain,NICD),随后将NICD转运至细胞核以调节下游基因表达。ITCH是一种属于HECT(homologous to E6-AP carboxyl terminus)家族的泛素连接酶,通过特异性激活Notch1信号传导的泛素,并促进NICD的泛素依赖性蛋白酶体降解而对Notch1信号进行负调节。阻断ITCH和衔接蛋白NUMB后,HBV cccDNA增加。cAMP反应元件结合蛋白(cAMP response element-binding protein,CREB)介导HBV cccDNA的转录,Notch激活CREB/CBP(CREB-binding protein)对HBV cccDNA的产生起关键作用,进而触发了HBV转录的激活,上调泛素连接酶ITCH-NUMB,以泛素依赖性蛋白酶体介导的方式阻断该途径,可明显抑制HBV cccDNA[42]。
5.3 HBx与HBV cccDNA转录
肝细胞核内HBV cccDNA的持续存在,是HBV感染慢性化、抗病毒治疗无法获得根治,以及停药后肝炎复发的主要原因。因此,HBV cccDNA持续沉默是慢性乙型肝炎“功能性治愈”的目标。病毒蛋白HBx对HBV cccDNA的转录过程发挥了关键作用。Cheng等[43]以HBx为靶标,通过HiBiT-tagged HBx蛋白检测系统,寻找到一个靶向作用于HBx的小分子化合物——双香豆素。在HBV感染细胞模型和人源化肝脏小鼠模型中,双香豆素均可显著促进HBx蛋白降解,进而显著抑制cccDNA的转录。进一步研究发现,双香豆素可作为细胞烟酰胺腺嘌呤双核苷酸磷酸盐(nicotinamide adenine dinucleotide phosphate,NADPH)醌氧化还原酶抑制剂,解除NADPH醌氧化还原酶对HBx蛋白的保护作用,从而促进HBx蛋白经泛素非依赖的20S蛋白酶体途径降解。
HBx与CRL4的相互作用对激活cccDNA基因组的表达起重要的作用。利用底物捕获蛋白质组学方法,证实染色体结构维持复合物(structural maintenance of chromosome,SMC)蛋白SMC5和SMC6是CRL4HBx的作用底物。在人肝细胞和人源化小鼠体内,HBx表达和HBV感染对SMC5/6复合物有降解作用,后者通过抑制HBV的基因表达以限制HBV复制[44]。HBx靶向SMC5/6是通过CRL4HBx连接酶泛素化及其后的蛋白酶体降解完成的。SMC5/6基因敲除可以恢复HBx缺陷型人肝细胞中HBV的复制[45]。泛素蛋白酶体系统在HBV生命周期中的更多调节作用详见图 3[39]。
6. 总结与展望
总之,HBV及其相关蛋白的泛素化研究越来越受到重视。HBsAg、HBeAg或HBV颗粒已被证明能够抑制Toll样受体诱导的抗病毒活性,降低IFNβ及IFN刺激基因的表达。病毒蛋白的降解伴随着体液和细胞免疫功能的恢复,对于控制和清除HBV感染具有重要意义。HBsAg的泛素化研究大多局限于蛋白相互作用及蛋白酶体降解,深层次的研究还相当缺乏。C基因相关蛋白的泛素化研究大多集中在构建泛素相关融合蛋白,以此增强体液和细胞免疫应答,达到抗病毒治疗的目的。P基因相关蛋白泛素化研究鉴定了多个与Pol蛋白降解有关的泛素连接酶。HBx是目前泛素化研究最多的一种HBV编码蛋白,它是蛋白酶体抑制剂,也是一种去泛素化酶,因此具有多重身份和功能。近年来,与HBV cccDNA相关的泛素化研究也逐渐增加,表明为了治愈慢性HBV感染,研究的领域在不断地拓展。相信为了实现世界卫生组织2030年消除HBV感染的目标,HBV及其相关蛋白质泛素化研究将成为今后关注和研究的一个热点。
-
[1] TONG S, REVILL P. Overview of hepatitis B viral replication and genetic variability[J]. J Hepatol, 2016, 64(1 Suppl): s4-s16. DOI: 10.1016/j.jhep.2016.01.027. [2] HICKE L. Protein regulation by monoubiquitin[J]. Nat Rev Mol Cell Biol, 2001, 2(3): 195-201. DOI: 10.1038/35056583. [3] HERSHKO A, CIECHANOVER A. The ubiquitin system[J]. Annu Rev Biochem, 1998, 67: 425-479. DOI: 10.1146/annurev.biochem.67.1.425. [4] ZHENG Y, GAO C. Fine-tuning of antiviral innate immunity by ubiquitination[J]. Adv Immunol, 2020, 145: 95-128. DOI: 10.1016/bs.ai.2019.11.004. [5] TOH QC, TAN TL, TEO WQ, et al. Identification of cellular membrane proteins interacting with hepatitis B surface antigen using yeast split-ubiquitin system[J]. Int J Med Sci, 2005, 2(3): 114-117. DOI: 10.7150/ijms.2.114. [6] HARTMANN-STVHLER C, PRANGE R. Hepatitis B virus large envelope protein interacts with gamma2-adaptin, a clathrin adaptor-related protein[J]. J Virol, 2001, 75(11): 5343-5351. DOI: 10.1128/JVI.75.11.5343-5351.2001. [7] MANGOLD CM, STREECK RE. Mutational analysis of the cysteine residues in the hepatitis B virus small envelope protein[J]. J Virol, 1993, 67(8): 4588-4597. DOI: 10.1128/JVI.67.8.4588-4597.1993. [8] LIU Y, TESTA JS, PHILIP R, et al. A ubiquitin independent degradation pathway utilized by a hepatitis B virus envelope protein to limit antigen presentation[J]. PLoS One, 2011, 6(9): e24477. DOI: 10.1371/journal.pone.0024477. [9] SIMSEK E, MEHTA A, ZHOU T, et al. Hepatitis B virus large and middle glycoproteins are degraded by a proteasome pathway in glucosidase-inhibited cells but not in cells with functional glucosidase enzyme[J]. J Virol, 2005, 79(20): 12914-12920. DOI: 10.1128/JVI.79.20.12914-12920.2005. [10] ROST M, MANN S, LAMBERT C, et al. Gamma-adaptin, a novel ubiquitin-interacting adaptor, and Nedd4 ubiquitin ligase control hepatitis B virus maturation[J]. J Biol Chem, 2006, 281(39): 29297-29308. DOI: 10.1074/jbc.M603517200. [11] GARCIA ML, BYFIELD R, ROBEK MD. Hepatitis B virus replication and release are independent of core lysine ubiquitination[J]. J Virol, 2009, 83(10): 4923-4933. DOI: 10.1128/JVI.02644-08. [12] LANGEROVÁ H, LUBYOVÁ B, ZÁBRANSKÝ A, et al. Hepatitis B core protein is post-translationally modified through K29-linked ubiquitination[J]. Cells, 2020, 9(12): 2547. DOI: 10.3390/cells9122547. [13] QIAN G, JIN F, CHANG L, et al. NIRF, a novel ubiquitin ligase, interacts with hepatitis B virus core protein and promotes its degradation[J]. Biotechnol Lett, 2012, 34(1): 29-36. DOI: 10.1007/s10529-011-0751-0. [14] QIAN G, HU B, ZHOU D, et al. NIRF, a novel ubiquitin ligase, inhibits hepatitis B virus replication through effect on HBV core protein and H3 histones[J]. DNA Cell Biol, 2015, 34(5): 327-332. DOI: 10.1089/dna.2014.2714. [15] CHEN JH, YU YS, LIU HH, et al. Ubiquitin conjugation of hepatitis B virus core antigen DNA vaccine leads to enhanced cell-mediated immune response in BALB/c mice[J]. Hepat Mon, 2011, 11(8): 620-628. DOI: 10.5812/kowsar.1735143x.689. [16] DAI SL, QIU CY, YAO J, et al. Packaging and identification of ubiquitinated HBcAg fusion gene and Tapasin gene expression recombinant lentivirus vectors[J]. China Med Herald, 2019, 16(4): 9-12, 17. https://www.cnki.com.cn/Article/CJFDTOTAL-YYCY201904003.htm戴胜兰, 邱彩玉, 姚俊, 等. 重组表达泛素化HBcAg融合基因和Tapasin基因的慢病毒的包装及鉴定[J]. 中国医药导报, 2019, 16(4): 9-12, 17. https://www.cnki.com.cn/Article/CJFDTOTAL-YYCY201904003.htm [17] CHEN JH, YU YS, CHEN XH, et al. Enhancement of CTLs induced by DCs loaded with ubiquitinated hepatitis B virus core antigen[J]. World J Gastroenterol, 2012, 18(12): 1319-1327. DOI: 10.3748/wjg.v18.i12.1319. [18] SONG L, ZHUO M, TANG Y, et al. Ubiquitin-modified hepatitis B virus core antigen effectively facilitates antigen presentation and enhances cytotoxic T lymphocyte activity via the cytoplasmic transduction peptide in vitro[J]. Mol Med Rep, 2015, 12(1): 289-296. DOI: 10.3892/mmr.2015.3352. [19] ZHUO M, SONG L, TANG Y, et al. Vaccination with ubiquitin-hepatitis B core antigen-cytoplasmic transduction peptide enhances the hepatitis B virus-specific cytotoxic T-lymphocyte immune response and inhibits hepatitis B virus replication in transgenic mice[J]. Mol Med Rep, 2015, 12(3): 3591-3598. DOI: 10.3892/mmr.2015.3834. [20] SONG L, ZHUO M, TANG Y, et al. Ubiquitin-hepatitis B core antigen-cytoplasmic transduction peptide enhances HBV-specific humoral and CTL immune responses in vivo[J]. Int Immunopharmacol, 2014, 23(1): 1-7. DOI: 10.1016/j.intimp.2014.08.006. [21] WANG Y, CUI L, YANG G, et al. Hepatitis B e antigen inhibits NF-κB activity by interrupting K63-linked ubiquitination of NEMO[J]. J Virol, 2019, 93(2): e00667-18. DOI: 10.1128/JVI.00667-18. [22] HOU L, ZHAO J, GAO S, et al. Restriction of hepatitis B virus replication by c-Abl-induced proteasomal degradation of the viral polymerase[J]. Sci Adv, 2019, 5(2): eaau7130. DOI: 10.1126/sciadv.aau7130. [23] LIU X, SHAO J, XIONG W, et al. Cellular cIAP2 gene expression associated with anti-HBV activity of TNF-alpha in hepatoblastoma cells[J]. J Interferon Cytokine Res, 2005, 25(10): 617-626. DOI: 10.1089/jir.2005.25.617. [24] WANG Z, NI J, LI J, et al. Inhibition of hepatitis B virus replication by cIAP2 involves accelerating the ubiquitin-proteasome-mediated destruction of polymerase[J]. J Virol, 2011, 85(21): 11457-11467. DOI: 10.1128/JVI.00879-11. [25] MU T, ZHAO X, ZHU Y, et al. The E3 ubiquitin ligase TRIM21 promotes HBV DNA polymerase degradation[J]. Viruses, 2020, 12(3): 346. DOI: 10.3390/v12030346. [26] LIU Y, LI J, CHEN J, et al. Hepatitis B virus polymerase disrupts K63-linked ubiquitination of STING to block innate cytosolic DNA-sensing pathways[J]. J Virol, 2015, 89(4): 2287-2300. DOI: 10.1128/JVI.02760-14. [27] HU Z, ZHANG Z, DOO E, et al. Hepatitis B virus X protein is both a substrate and a potential inhibitor of the proteasome complex[J]. J Virol, 1999, 73(9): 7231-7240. DOI: 10.1128/JVI.73.9.7231-7240.1999. [28] CHEN H, ZHANG Y, YE S, et al. Chromatin remodelling factor BAF155 protects hepatitis B virus X protein (HBx) from ubiquitin-independent proteasomal degradation[J]. Emerg Microbes Infect, 2019, 8(1): 1393-1405. DOI: 10.1080/22221751.2019.1666661. [29] ZHANG JF, XIONG HL, CAO JL, et al. A cell-penetrating whole molecule antibody targeting intracellular HBx suppresses hepatitis B virus via TRIM21-dependent pathway[J]. Theranostics, 2018, 8(2): 549-562. DOI: 10.7150/thno.20047. [30] ZHAO J, WU J, CAI H, et al. E3 ubiquitin ligase Siah-1 is down-regulated and fails to target natural HBx truncates for degradation in hepatocellular carcinoma[J]. J Cancer, 2016, 7(4): 418-426. DOI: 10.7150/jca.13019. [31] YEOM S, KIM SS, JEONG H, et al. Hepatitis B virus X protein activates E3 ubiquitin ligase Siah-1 to control virus propagation via a negative feedback loop[J]. J Gen Virol, 2017, 98(7): 1774-1784. DOI: 10.1099/jgv.0.000856. [32] YOO YS, PARK YJ, LEE HS, et al. Mitochondria ubiquitin ligase, MARCH5 resolves hepatitis B virus X protein aggregates in the liver pathogenesis[J]. Cell Death Dis, 2019, 10(12): 938. DOI: 10.1038/s41419-019-2175-z. [33] ZHANG H, HUANG C, WANG Y, et al. Hepatitis B virus X protein sensitizes TRAIL-induced hepatocyte apoptosis by inhibiting the E3 ubiquitin ligase A20[J]. PLoS One, 2015, 10(5): e0127329. DOI: 10.1371/journal.pone.0127329. [34] MOLINA-JIMÉNEZ F, BENEDICTO I, MURATA M, et al. Expression of pituitary tumor-transforming gene 1 (PTTG1)/securin in hepatitis B virus (HBV)-associated liver diseases: Evidence for an HBV X protein-mediated inhibition of PTTG1 ubiquitination and degradation[J]. Hepatology, 2010, 51(3): 777-787. DOI: 10.1002/hep.23468. [35] LEE S, KIM W, KO C, et al. Hepatitis B virus X protein enhances Myc stability by inhibiting SCF(Skp2) ubiquitin E3 ligase-mediated Myc ubiquitination and contributes to oncogenesis[J]. Oncogene, 2016, 35(14): 1857-1867. DOI: 10.1038/onc.2015.251. [36] GUO L, WANG X, REN L, et al. HBx affects CUL4-DDB1 function in both positive and negative manners[J]. Biochem Biophys Res Commun, 2014, 450(4): 1492-1497. DOI: 10.1016/j.bbrc.2014.07.019. [37] JIANG J, TANG H. Mechanism of inhibiting type I interferon induction by hepatitis B virus X protein[J]. Protein Cell, 2010, 1(12): 1106-1117. DOI: 10.1007/s13238-010-0141-8. [38] WEI C, NI C, SONG T, et al. The hepatitis B virus X protein disrupts innate immunity by downregulating mitochondrial antiviral signaling protein[J]. J Immunol, 2010, 185(2): 1158-1168. DOI: 10.4049/jimmunol.0903874. [39] KONG F, YOU H, KONG D, et al. The interaction of hepatitis B virus with the ubiquitin proteasome system in viral replication and associated pathogenesis[J]. Virol J, 2019, 16(1): 73. DOI: 10.1186/s12985-019-1183-z. [40] CHEN J, WU M, LIU K, et al. New insights into hepatitis B virus biology and implications for novel antiviral strategies[J]. National Science Review, 2015, 2(4): 296-313. DOI: 10.1093/nsr/nwv044. [41] ZHOU L, REN JH, CHENG ST, et al. A functional variant in ubiquitin conjugating enzyme E2 L3 contributes to hepatitis B virus infection and maintains covalently closed circular DNA stability by inducing degradation of apolipoprotein B mRNA editing enzyme catalytic subunit 3A[J]. Hepatology, 2019, 69(5): 1885-1902. DOI: 10.1002/hep.30497. [42] WANG Z, KAWAGUCHI K, HONDA M, et al. Notch signaling facilitates hepatitis B virus covalently closed circular DNA transcription via cAMP response element-binding protein with E3 ubiquitin ligase-modulation[J]. Sci Rep, 2019, 9(1): 1621. DOI: 10.1038/s41598-018-38139-5. [43] CHENG ST, HU JL, REN JH, et al. Dicoumarol, an NQO1 inhibitor, blocks cccDNA transcription by promoting degradation of HBx[J]. J Hepatol, 2021, 74(3): 522-534. DOI: 10.1016/j.jhep.2020.09.019. [44] DECORSIÈRE A, MUELLER H, van BREUGEL PC, et al. Hepatitis B virus X protein identifies the Smc5/6 complex as a host restriction factor[J]. Nature, 2016, 531(7594): 386-389. DOI: 10.1038/nature17170. [45] MURPHY CM, XU Y, LI F, et al. Hepatitis B virus X protein promotes degradation of SMC5/6 to enhance HBV replication[J]. Cell Rep, 2016, 16(11): 2846-2854. DOI: 10.1016/j.celrep.2016.08.026. 期刊类型引用(1)
1. 陈为,沈楠,韩宛娜,郗艳丽,任旷,金连海,许娜. Toll样受体4接头分子对乳酸诱导巨噬细胞M2极化的促进作用及其机制. 吉林大学学报(医学版). 2022(05): 1190-1199 .  百度学术
百度学术其他类型引用(0)
-


 下载:
下载:



 下载:
下载:

 百度学术
百度学术

