
 PDF下载 ( 1016 KB)
PDF下载 ( 1016 KB)

原发性胆汁性胆管炎的病理学诊断
DOI: 10.12449/JCH240604
利益冲突声明:所有作者均声明不存在利益冲突。
作者贡献声明:王秋鹏负责撰写修改论文;甘梅富负责拟定写作思路,指导撰写文章并最后定稿。
-
摘要: 近年来原发性胆汁性胆管炎发病机制、诊断及治疗等方面的研究均取得新的进展。根据患者临床特征及病理学特点准确诊断、评估预后及风险分层对原发性胆汁性胆管炎的治疗具有十分重要的意义。本文就原发性胆汁性胆管炎的临床病理特征及治疗进展做一概述。Abstract: In recent years, new advances have been achieved for the research on the pathogenesis, diagnosis, and treatment of primary biliary cholangitis (PBC). Accurate diagnosis, prognosis assessment, and risk stratification based on the clinicopathological features of patients are important for the treatment of PBC. This article summarizes the advances in the clinicopathological features and treatment of PBC.
-
Key words:
- Primary Biliary Cholangitis /
- Biopsy /
- Pathology /
- Diagnosis
-
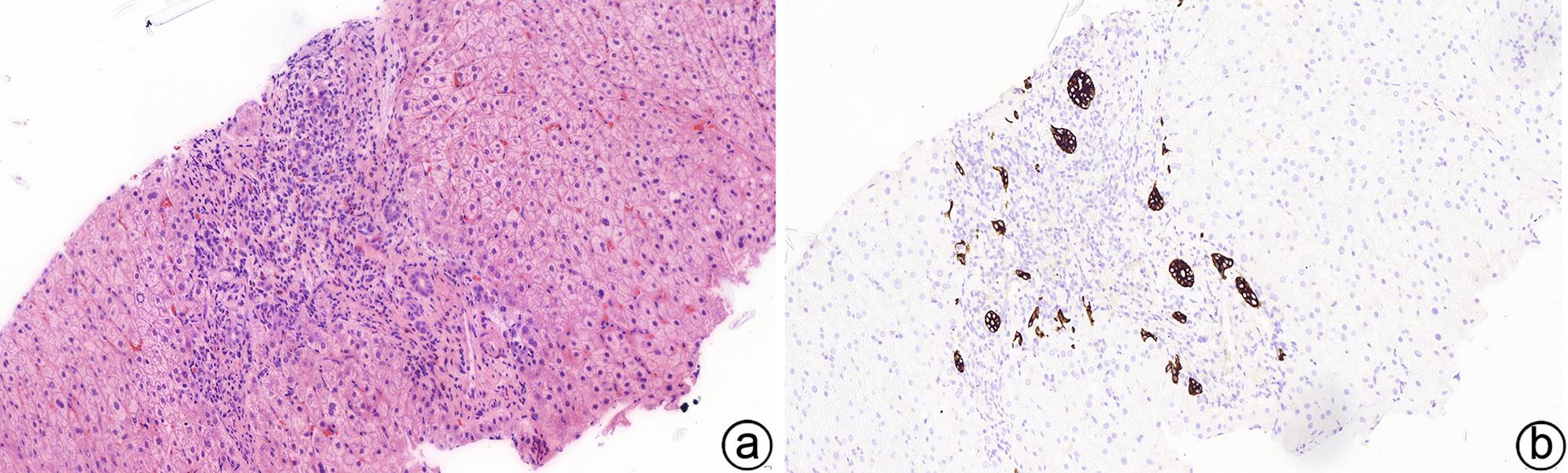
注: a,汇管区周边细胆管增生,可见界板性肝炎及炎性纤维间隔(HE染色,×20);b,CK7免疫组化染色显示反应性增生的细胆管(EnVision法,×20)。
图 2 PBC Ⅱ期
Figure 2. Stage 2 PBC
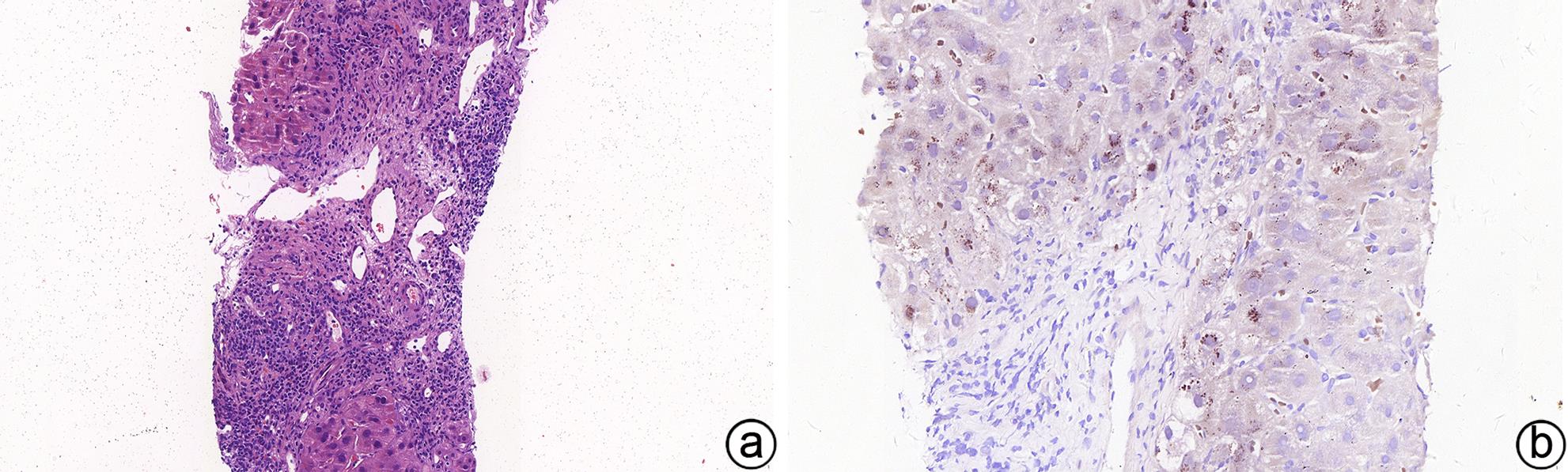
注: a,汇管区固有小胆管缺失,汇管区及间隔周围界面炎范围扩大,可见桥接性纤维化(HE染色,×20);b,汇管区周边肝细胞内铜颗粒沉积(罗丹宁染色,×40)。
图 3 PBC Ⅲ期
Figure 3. Stage 3 PBC

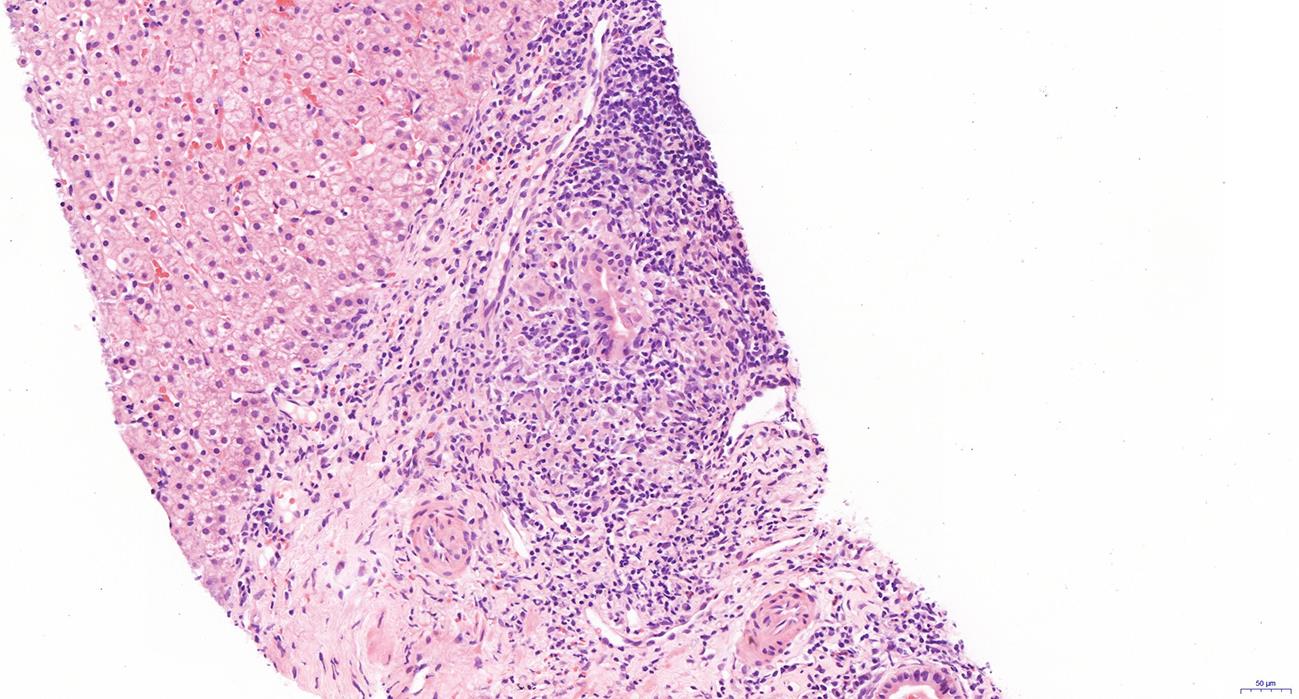
注: 汇管区周边肝细胞体积增大,胞质疏松,呈羽毛状变性,形成汇管区-汇管桥接性纤维化(HE染色,×20)。
图 4 PBC Ⅳ期
Figure 4. Stage 4 PBC
-
[1] SMYK DS, RIGOPOULOU EI, LLEO A, et al. Immunopathogenesis of primary biliary cirrhosis: an old wives’ tale[J]. Immun Ageing, 2011, 8( 1): 12. DOI: 10.1186/1742-4933-8-12. [2] KIM WR, LINDOR KD, LOCKE GR 3rd, et al. Epidemiology and natural history of primary biliary cirrhosis in a US community[J]. Gastroenterology, 2000, 119( 6): 1631- 1636. DOI: 10.1053/gast.2000.20197. [3] WATSON RG, ANGUS PW, DEWAR M, et al. Low prevalence of primary biliary cirrhosis in Victoria, Australia. Melbourne Liver Group[J]. Gut, 1995, 36( 6): 927- 930. DOI: 10.1136/gut.36.6.927. [4] TANAKA A, MA X, YOKOSUKA O, et al. Autoimmune liver diseases in the Asia-Pacific region: Proceedings of APASL symposium on AIH and PBC 2016[J]. Hepatol Int, 2016, 10( 6): 909- 915. DOI: 10.1007/s12072-016-9767-9. [5] LLEO A, JEPSEN P, MORENGHI E, et al. Evolving trends in female to male incidence and male mortality of primary biliary cholangitis[J]. Sci Rep, 2016, 6: 25906. DOI: 10.1038/srep25906. [6] JOHN BV, AITCHESON G, SCHWARTZ KB, et al. Male sex is associated with higher rates of liver-related mortality in primary biliary cholangitis and cirrhosis[J]. Hepatology, 2021, 74( 2): 879- 891. DOI: 10.1002/hep.31776. [7] FAN XL, WANG TT, SHEN Y, et al. Underestimated male prevalence of primary biliary cholangitis in China: Results of a 16-yr cohort study involving 769 patients[J]. Sci Rep, 2017, 7: 6560. DOI: 10.1038/s41598-017-06807-7. [8] GERSHWIN ME, SELMI C, WORMAN HJ, et al. Risk factors and comorbidities in primary biliary cirrhosis: a controlled interview-based study of 1032 patients[J]. Hepatology, 2005, 42( 5): 1194- 202. DOI: 10.1002/hep.20907. [9] LEUNG PS, WANG J, NAIYANETR P, et al. Environment and primary biliary cirrhosis: electrophilic drugs and the induction of AMA[J]. J Autoimmun, 2013, 41: 79- 86. DOI: 10.1016/j.jaut.2012.12.007. [10] TANAKA A, LEUNG PSC, GERSHWIN ME. Pathogen infections and primary biliary cholangitis[J]. Clin Exp Immunol, 2019, 195( 1): 25- 34. DOI: 10.1111/cei.13198. [11] SHIMODA S, NAKAMURA M, ISHIBASHI H, et al. Molecular mimicry of mitochondrial and nuclear autoantigens in primary biliary cirrhosis[J]. Gastroenterology, 2003, 124( 7): 1915- 1925. DOI: 10.1016/s0016-5085(03)00387-1. [12] FLOREANI A, NACCARATO R, CHIARAMONTE M. Prevalence of familial disease in primary biliary cirrhosis in Italy[J]. J Hepatol, 1997, 26( 3): 737- 738. DOI: 10.1016/s0168-8278(97)80444-8. [13] JONES DE, WATT FE, METCALF JV, et al. Familial primary biliary cirrhosis reassessed: A geographically-based population study[J]. J Hepatol, 1999, 30( 3): 402- 407. DOI: 10.1016/s0168-8278(99)80097-x. [14] TSUCHIYA N, OHASHI J. Human immune system diversity and its implications in diseases[J]. J Hum Genet, 2015, 60( 11): 655- 656. DOI: 10.1038/jhg.2015.101. [15] NAKAMURA M, YASUNAMI M, KONDO H, et al. Analysis of HLA-DRB1 polymorphisms in Japanese patients with primary biliary cirrhosis(PBC): The HLA-DRB1polymorphism determines the relative risk of antinuclear antibodies for disease progression in PBC[J]. Hepatol Res, 2010, 40( 5): 494- 504. DOI: 10.1111/j.1872-034X.2010.00631.x. [16] UMEMURA T, JOSHITA S, ICHIJO T, et al. Human leukocyte antigen class II molecules confer both susceptibility and progression in Japanese patients with primary biliary cirrhosis[J]. Hepatology, 2012, 55( 2): 506- 511. DOI: 10.1002/hep.24705. [17] ZHAO DT, LIAO HY, ZHANG X, et al. Human leucocyte antigen alleles and haplotypes and their associations with antinuclear antibodies features in Chinese patients with primary biliary cirrhosis[J]. Liver Int, 2014, 34( 2): 220- 226. DOI: 10.1111/liv.12236. [18] YASUNAMI M, NAKAMURA H, TOKUNAGA K, et al. Principal contribution of HLA-DQ alleles, DQB1*06:04 and DQB1*03:01, to disease resistance against primary biliary cholangitis in a Japanese population[J]. Sci Rep, 2017, 7( 1): 11093. DOI: 10.1038/s41598-017-11148-6. [19] WANG C, ZHENG XD, TANG RQ, et al. Fine mapping of the MHC region identifies major independent variants associated with Han Chinese primary biliary cholangitis[J]. J Autoimmun, 2020, 107: 102372. DOI: 10.1016/j.jaut.2019.102372. [20] CORDELL HJ, FRYETT JJ, UENO K, et al. An international genome-wide meta-analysis of primary biliary cholangitis: Novel risk loci and candidate drugs[J]. J Hepatol, 2021, 75( 3): 572- 581. DOI: 10.1016/j.jhep.2021.04.055. [21] ASSELTA R, PARABOSCHI EM, GERUSSI A, et al. X chromosome contribution to the genetic architecture of primary biliary cholangitis[J]. Gastroenterology, 2021, 160( 7): 2483- 2495. e 26. DOI: 10.1053/j.gastro.2021.02.061. [22] HITOMI Y, NAKAMURA M. The genetics of primary biliary cholangitis: A GWAS and Post-GWAS update[J]. Genes(Basel), 2023, 14( 2): 405. DOI: 10.3390/genes14020405. [23] OKADA Y, WU D, TRYNKA G, et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery[J]. Nature, 2014, 506( 7488): 376- 381. DOI: 10.1038/nature12873. [24] ZHANG JZ, JIANG KW, LV L, et al. Use of genome-wide association studies for cancer research and drug repositioning[J]. PLoS One, 2015, 10( 3): e0116477. DOI: 10.1371/journal.pone.0116477. [25] IMAMURA M, TAKAHASHI A, YAMAUCHI T, et al. Genome-wide association studies in the Japanese population identify seven novel loci for type 2 diabetes[J]. Nat Commun, 2016, 7: 10531. DOI: 10.1038/ncomms10531. [26] POUPON RE, CHRÉTIEN Y, CHAZOUILLÈRES O, et al. Quality of life in patients with primary biliary cirrhosis[J]. Hepatology, 2004, 40( 2): 489- 494. DOI: 10.1002/hep.20276. [27] FILLER K, LYON D, BENNETT J, et al. Association of mitochondrial dysfunction and fatigue: A review of the literature[J]. BBA Clin, 2014, 1: 12- 23. DOI: 10.1016/j.bbacli.2014.04.001. [28] TALWALKAR JA, SOUTO E, JORGENSEN RA, et al. Natural history of pruritus in primary biliary cirrhosis[J]. Clin Gastroenterol Hepatol, 2003, 1( 4): 297- 302. [29] PHILLIPS JR, ANGULO P, PETTERSON T, et al. Fat-soluble vitamin levels in patients with primary biliary cirrhosis[J]. Am J Gastroenterol, 2001, 96( 9): 2745- 2750. DOI: 10.1111/j.1572-0241.2001.04134.x. [30] KAPLAN MM, GERSHWIN ME. Primary biliary cirrhosis[J]. N Engl J Med, 2005, 353( 12): 1261- 1273. DOI: 10.1056/NEJMra043898. [31] BIZZARO N, COVINI G, ROSINA F, et al. Overcoming a“probable” diagnosis in antimitochondrial antibody negative primary biliary cirrhosis: Study of 100 sera and review of the literature[J]. Clin Rev Allergy Immunol, 2012, 42( 3): 288- 297. DOI: 10.1007/s12016-010-8234-y. [32] MATTALIA A, QUARANTA S, LEUNG PSC, et al. Characterization of antimitochondrial antibodies in healthy adults[J]. Hepatology, 1998, 27( 3): 656- 661. DOI: 10.1002/hep.510270303. [33] SHIBATA M, ONOZUKA Y, MORIZANE T, et al. Prevalence of antimitochondrial antibody in Japanese corporate workers in Kanagawa prefecture[J]. J Gastroenterol, 2004, 39( 3): 255- 259. DOI: 10.1007/s00535-003-1285-6. [34] DAHLQVIST G, GAOUAR F, CARRAT F, et al. Large-scale characterization study of patients with antimitochondrial antibodies but nonestablished primary biliary cholangitis[J]. Hepatology, 2017, 65( 1): 152- 163. DOI: 10.1002/hep.28859. [35] YANG F, YANG Y, WANG Q, et al. The risk predictive values of UK-PBC and GLOBE scoring system in Chinese patients with primary biliary cholangitis: the additional effect of anti-gp210[J]. Aliment Pharmacol Ther, 2017, 45( 5): 733- 743. DOI: 10.1111/apt.13927. [36] European Association for the Study of the Liver. EASL clinical practice guidelines: The diagnosis and management of patients with primary biliary cholangitis[J]. J Hepatol, 2017, 67( 1): 145- 172. DOI: 10.1016/j.jhep.2017.03.022. [37] CHAZOUILLÈRES O, WENDUM D, SERFATY L, et al. Primary biliary cirrhosis-autoimmune hepatitis overlap syndrome: Clinical features and response to therapy[J]. Hepatology, 1998, 28( 2): 296- 301. DOI: 10.1002/hep.510280203. [38] Chinese Society of Hepatology, Chinese Medical Association. Guidelines on the diagnosis and management of primary biliary cholangitis(2021)[J]. J Clin Hepatol, 2022, 38( 1): 35- 41. DOI: 10.3969/j.issn.1001-5256.2022.01.007.中华医学会肝病学分会. 原发性胆汁性胆管炎的诊断和治疗指南(2021)[J]. 临床肝胆病杂志, 2022, 38( 1): 35- 41. DOI: 10.3969/j.issn.1001-5256.2022.01.007. -

 下载:
下载:

 本文二维码
本文二维码
图(5)
计量
- 文章访问数: 1765
- HTML全文浏览量: 1083
- PDF下载量: 218
- 被引次数: 0

