
 PDF下载 ( 1291 KB)
PDF下载 ( 1291 KB)

-
摘要:
自身免疫性肝炎(AIH)是免疫介导的肝实质炎症性疾病,以高丙种球蛋白血症、自身抗体阳性和典型的肝组织学异常为特征。然而,AIH临床表现多样,缺乏特异性的血清学标志物,因此AIH的诊断困难,具有挑战性。虽然汇管区淋巴浆细胞浸润、界面性肝炎、淋巴细胞穿入现象和肝细胞玫瑰花结为AIH典型的组织学特征,但在AIH中尚可观察到其他的一些形态学改变,包括小叶中心性坏死、Kupffer细胞内透明小球等。所以没有一个单一的组织学特征可直接诊断AIH,需结合临床和实验室检查,并排除其他原因引起的肝脏疾病,方可做出正确的诊断。本文总结了AIH的组织学特征、不同的病理组织学谱、常见的临床问题、主要的鉴别诊断和最新进展。
Abstract:Autoimmune hepatitis (AIH) is an immune-mediated inflammatory disease of the liver parenchyma, which is characterized by hypergammaglobulinemia, the presence of autoantibodies, and typical abnormalities in liver histology; however, the diverse clinical manifestations of AIH and the lack of specific serological markers have brought difficulties and challenges in the diagnosis of AIH. Although portal lymphoplasmacytic infiltration, interface hepatitis, lymphocyte emperipolesis, and hepatocyte rosettes are the typical histological features of AIH, many other histological features can also be observed in AIH, including centrilobular necrosis and Kupffer cell hyaline globules. Therefore, no single histological feature can be used for the diagnosis of AIH, and a confirmed diagnosis should be made with reference to clinical and laboratory examinations, with the exclusion of liver diseases due to other causes. This article summarizes the histological features of AIH, different histopathological spectrum, common clinical issues, differential diagnosis, and recent advances.
-
Key words:
- Hepatitis, Autoimmune /
- Biopsy /
- Pathology /
- Diagnosis
-
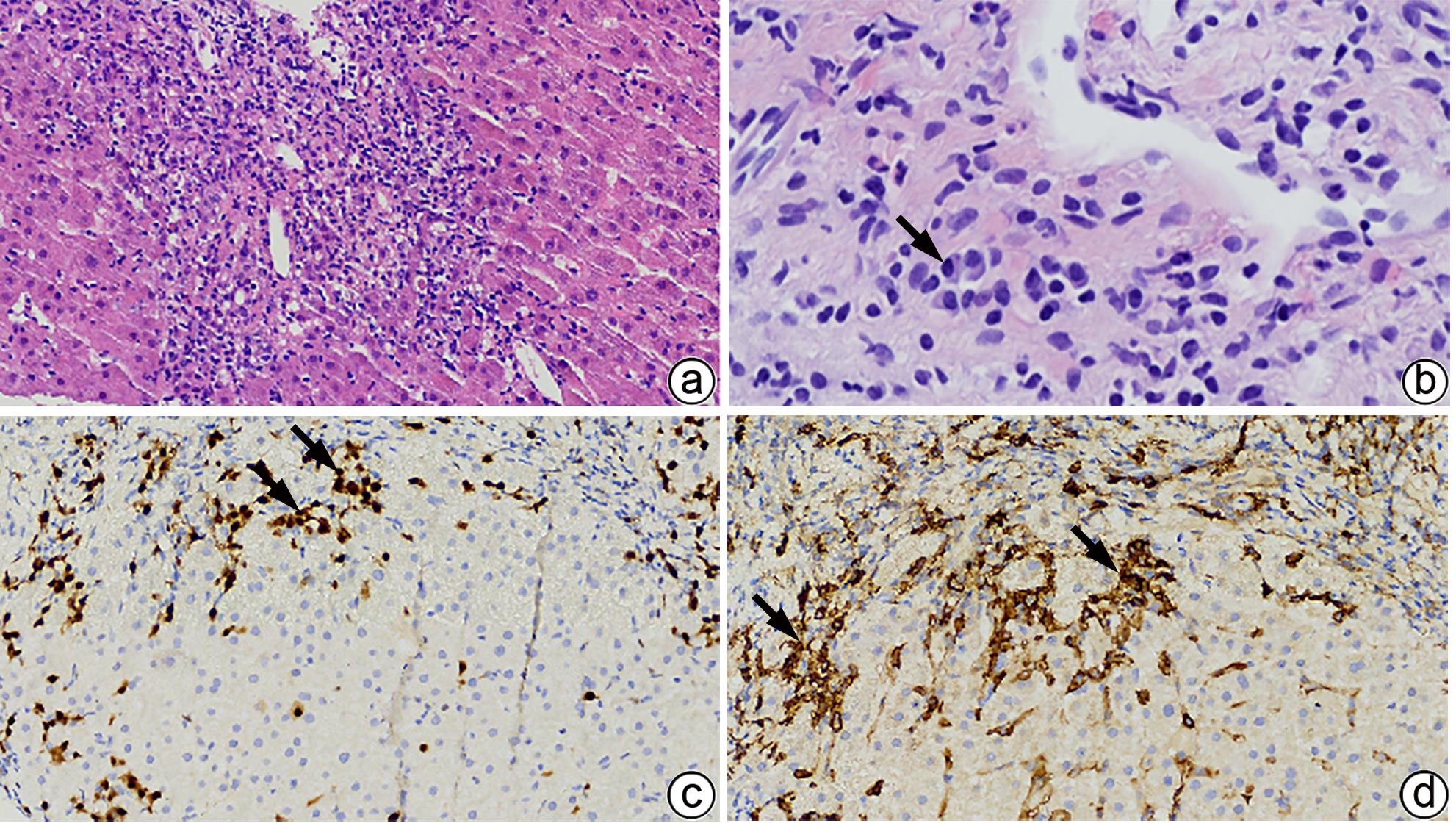
注: a,汇管区淋巴浆细胞浸润,界面性肝炎(HE染色,×10)b,汇管区簇状浆细胞浸润(箭头)(HE染色,×40);c,簇状浆细胞阳性(箭头)(MUM-1染色,×20);d,簇状浆细胞阳性(箭头)(CD38染色,×20)。
图 1 汇管区淋巴浆细胞浸润和界面性肝炎
Figure 1. Portal lymphoplasmacytic infiltrate and interface hepatitis

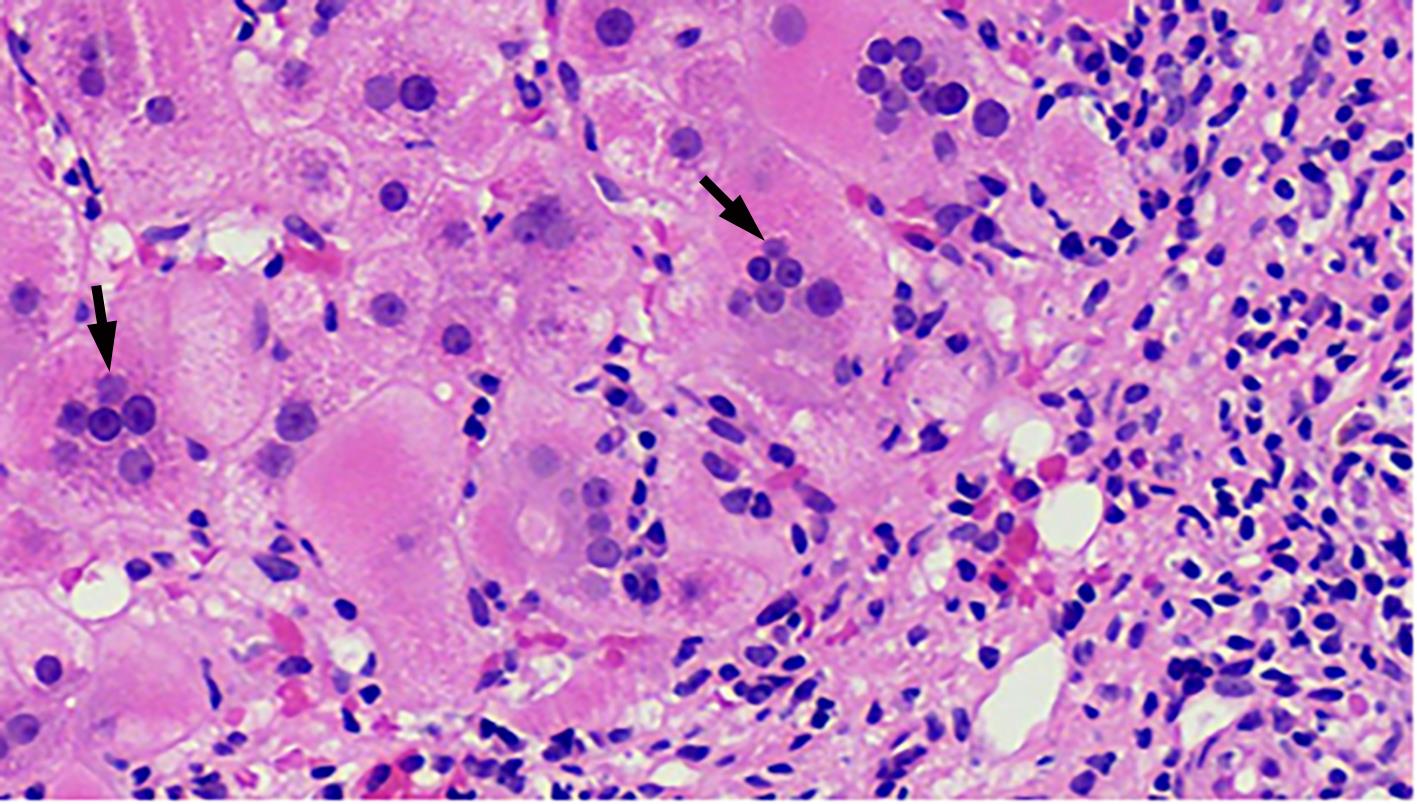
注: 箭头处为小叶中心性坏死。
图 4 小叶中心性坏死(HE染色,×10)
Figure 4. Centrilobular necrosis(HE staining,×10)
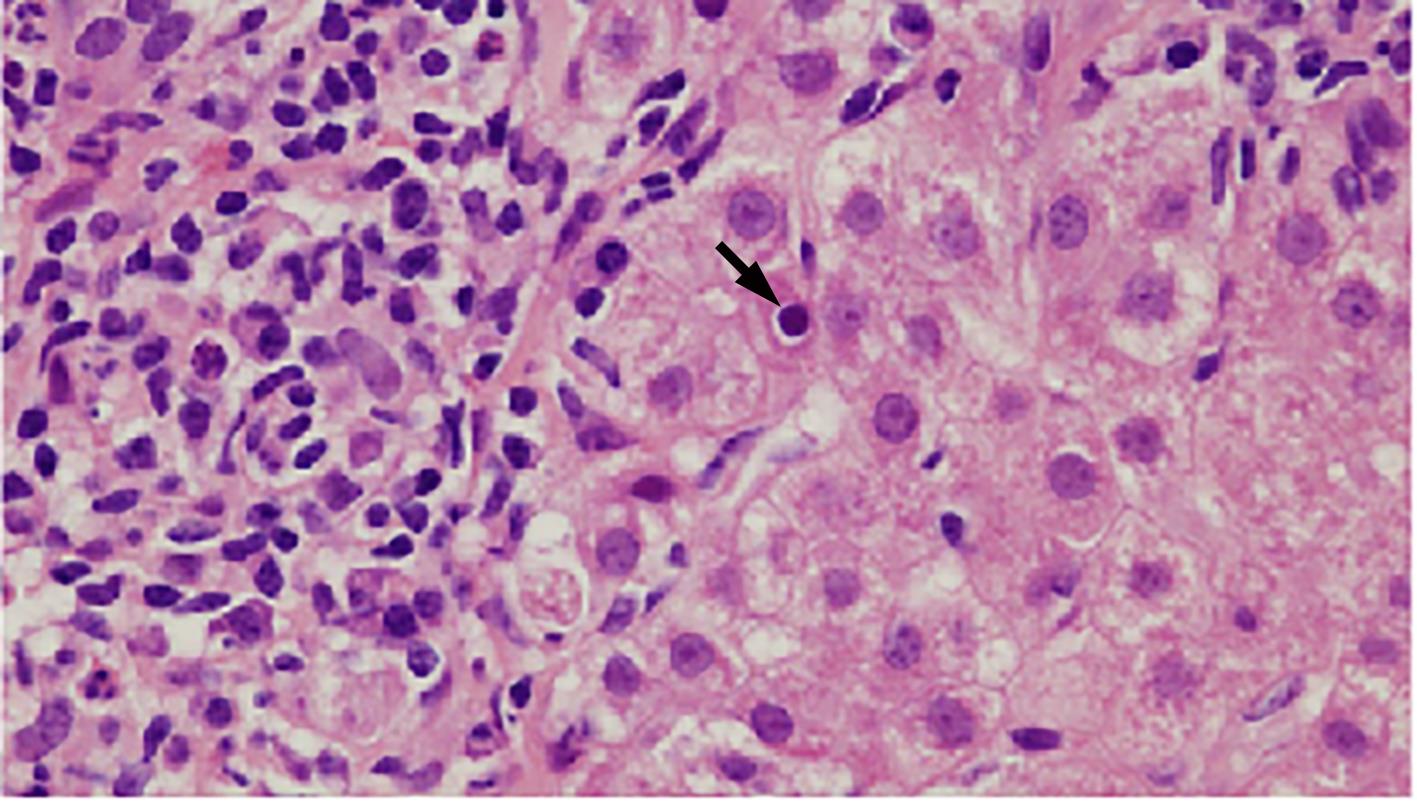
注: 箭头处为合胞体多核巨肝细胞。
图 5 合胞体多核巨肝细胞(HE染色,×20)
Figure 5. Syncytial multinucleated giant cells(HE staining,×20)
表 1 基于汇管区和小叶炎症的AIH诊断标准
Table 1. AIH diagnostic criteria based on portal area and lobular inflammation
诊断 汇管区炎症 小叶炎症 极可能 (likely AIH) 汇管区淋巴、浆细胞浸润+ 以下1种或2种特征: (1)轻度以上界面性肝炎, (2)轻度以上小叶炎; 缺乏提示其他肝病的组织学形态 轻度以上小叶炎(伴或不伴小叶中心性坏死)+至少1种以下特征: (1)淋巴、浆细胞浸润, (2)界面性肝炎, (3)汇管区纤维化; 缺乏提示其他肝病的组织学形态 可能 (possible AIH) 汇管区淋巴、浆细胞浸润; 不具有上述极可能的任一特征; 缺乏提示其他肝病的组织学形态或 伴上述1种或2种极可能的特征; 出现提示其他肝病的组织学形态 小叶性肝炎,伴或不伴小叶中心性坏死; 不具有上述极可能的任一特征; 缺乏提示其他肝病的组织学形态或 伴上述任一极可能的特征; 出现提示其他肝病的组织学形态 不太可能 (likely AIH) 汇管区炎症; 不具有上述极可能的任一特征; 出现提示其他肝病的组织学形态 任何程度的小叶性肝炎; 不具有上述极可能的任一特征; 出现提示其他肝病的组织学形态  下载: 导出CSV
下载: 导出CSV
-
[1] ZHANG XC, JAIN D. The many faces and pathologic diagnostic challenges of autoimmune hepatitis[J]. Hum Pathol, 2023, 132: 114- 125. DOI: 10.1016/j.humpath.2022.06.019. [2] MANNS MP, CZAJA AJ, GORHAM JD, et al. Diagnosis and management of autoimmune hepatitis[J]. Hepatology, 2010, 51( 6): 2193- 2213. DOI: 10.1002/hep.23584. [3] MACK CL, ADAMS D, ASSIS DN, et al. Diagnosis and management of autoimmune hepatitis in adults and children: 2019 practice guidance and guidelines from the American association for the study of liver diseases[J]. Hepatology, 2020, 72( 2): 671- 722. DOI: 10.1002/hep.31065. [4] SCIVERES M, NASTASIO S, MAGGIORE G. Novel diagnostic and therapeutic strategies in juvenile autoimmune hepatitis[J]. Front Pediatr, 2019, 7: 382. DOI: 10.3389/fped.2019.00382. [5] NASTASIO S, MOSCA A, ALTERIO T, et al. Juvenile autoimmune hepatitis: Recent advances in diagnosis, management and long-term outcome[J]. Diagnostics, 2023, 13( 17): 2753. DOI: 10.3390/diagnostics13172753. [6] GURUNG A, ASSIS DN, MCCARTY TR, et al. Histologic features of autoimmune hepatitis: A critical appraisal[J]. Hum Pathol, 2018, 82: 51- 60. DOI: 10.1016/j.humpath.2018.07.014. [7] FRANCESCHINI T, VASURI F, MURATORI P, et al. A practical histological approach to the diagnosis of autoimmune hepatitis: experience of an Italian tertiary referral center[J]. Virchows Arch, 2021, 479( 5): 937- 945. DOI: 10.1007/s00428-021-03122-5. [8] ZHAO SX, LI WC, FU N, et al. Emperipolesis mediated by CD8+ T cells correlates with biliary epithelia cell injury in primary biliary cholangitis[J]. J Cell Mol Med, 2020, 24( 2): 1268- 1275. DOI: 10.1111/jcmm.14752. [9] LOHSE AW, SEBODE M, BHATHAL PS, et al. Consensus recommendations for histological criteria of autoimmune hepatitis from the International AIH Pathology Group: Results of a workshop on AIH histology hosted by the European Reference Network on Hepatological Diseases and the European Society of Pathology: Results of a workshop on AIH histology hosted by the European Reference Network on Hepatological Diseases and the European Society of Pathology[J]. Liver Int, 2022, 42( 5): 1058- 1069. DOI: 10.1111/liv.15217. [10] AIZAWA Y, ABE H, SUGITA T, et al. Centrilobular zonal necrosis as a hallmark of a distinctive subtype of autoimmune hepatitis[J]. Eur J Gastroenterol Hepatol, 2016, 28( 4): 391- 397. DOI: 10.1097/MEG.0000000000000545. [11] UEDA K, AIZAWA Y, KINOSHITA C, et al. Centrilobular zonal necrosis is a unique subtype of autoimmune hepatitis: A cohort study[J]. Medicine, 2022, 101( 29): e29484. DOI: 10.1097/MD.0000000000029484. [12] CANH HN, HARADA K, OUCHI H, et al. Acute presentation of autoimmune hepatitis: A multicentre study with detailed histological evaluation in a large cohort of patients[J]. J Clin Pathol, 2017, 70( 11): 961- 969. DOI: 10.1136/jclinpath-2016-204271. [13] BIHARI C, RASTOGI A, SARIN SK. Postinfantile giant cell hepatitis: An etiological and prognostic perspective[J]. Hepat Res Treat, 2013, 2013: 601290. DOI: 10.1155/2013/601290. [14] KHEDR MA, ADAWY NM, SALIM TA, et al. Kupffer cell hyaline globules in children with autoimmune hepatitis[J]. J Clin Exp Hepatol, 2022, 12( 1): 20- 28. DOI: 10.1016/j.jceh.2021.04.013. [15] CZAJA AJ, CARPENTER HA. Autoimmune hepatitis with incidental histologic features of bile duct injury[J]. Hepatology, 2001, 34( 4 Pt 1): 659- 665. DOI: 10.1053/jhep.2001.27562. [16] MEHENDIRATTA V, MITROO P, BOMBONATI A, et al. Serologic markers do not predict histologic severity or response to treatment in patients with autoimmune hepatitis[J]. Clin Gastroenterol Hepatol, 2009, 7( 1): 98- 103. DOI: 10.1016/j.cgh.2008.08.043. [17] KHEDR MA, SALEM TA, BOGHDADI GM, et al. Seronegative autoimmune hepatitis in children: A real diagnostic challenge[J]. Wien Klin Wochenschr, 2022, 134( 5-6): 195- 201. DOI: 10.1007/s00508-021-01907-x. [18] HARTL J, MIQUEL R, ZACHOU K, et al. Features and outcome of AIH patients without elevation of IgG[J]. JHEP Rep, 2020, 2( 3): 100094. DOI: 10.1016/j.jhepr.2020.100094. [19] MONTANO-LOZA AJ, CARPENTER HA, CZAJA AJ. Frequency, behavior, and prognostic implications of antimitochondrial antibodies in type 1 autoimmune hepatitis[J]. J Clin Gastroenterol, 2008, 42( 9): 1047- 1053. DOI: 10.1097/MCG.0b013e3181587d18. [20] de ROZA MA, LAMBA M, GOH GBB, et al. Immunoglobulin G in non-alcoholic steatohepatitis predicts clinical outcome: A prospective multi-centre cohort study[J]. World J Gastroenterol, 2021, 27( 43): 7563- 7571. DOI: 10.3748/wjg.v27.i43.7563. [21] SUN SM, WANG YY, ZHANG Q, et al. Serum levels of immunoglobulins in an adult population and their relationship with nonalcoholic fatty liver disease[J]. J Dig Dis, 2018, 19( 8): 498- 507. DOI: 10.1111/1751-2980.12646. [22] TAKAHASHI A, ARINAGA-HINO T, OHIRA H, et al. Nonalcoholic fatty liver disease in patients with autoimmune hepatitis[J]. JGH Open, 2018, 2( 2): 54- 58. DOI: 10.1002/jgh3.12046. [23] JOHNSON PJ, MCFARLANE IG. Meeting report: International autoimmune hepatitis group[J]. Hepatology, 1993, 18( 4): 998- 1005. DOI: 10.1002/hep.1840180435. [24] ALVAREZ F, BERG PA, BIANCHI FB, et al. International Autoimmune Hepatitis Group Report: Review of criteria for diagnosis of autoimmune hepatitis[J]. J Hepatol, 1999, 31( 5): 929- 938. DOI: 10.1016/s0168-8278(99)80297-9. [25] HENNES EM, ZENIYA M, CZAJA AJ, et al. Simplified criteria for the diagnosis of autoimmune Hepatitis[J]. Hepatology, 2008, 48: 169- 176. DOI: 10.1002/hep.22322. [26] KABAÇAM G, WAHLIN S, EFE C. Autoimmune hepatitis triggered by COVID-19: A report of two cases[J]. Liver Int, 2021, 41( 10): 2527- 2528. DOI: 10.1111/liv.15044. [27] ŞENER AG, AYDıN N, CEYLAN C, et al. Investigation of antinuclear antibodies in chronic hepatitis B patients[J]. Mikrobiyol Bul, 2018, 52( 4): 425- 430. DOI: 10.5578/mb.67262. [28] ZHANG XC, OUYANG J, THUNG SN. Histopathologic manifestations of drug-induced hepatotoxicity[J]. Clin Liver Dis, 2013, 17( 4): 547- 564, vii-viii. DOI: 10.1016/j.cld.2013.07.004. [29] PATIL PA, ZHANG XC. Pathologic manifestations of gastrointestinal and hepatobiliary injury in immune checkpoint inhibitor therapy[J]. Arch Pathol Lab Med, 2021, 145( 5): 571- 582. DOI: 10.5858/arpa.2020-0070-RA. [30] SUZUKI A, BRUNT EM, KLEINER DE, et al. The use of liver biopsy evaluation in discrimination of idiopathic autoimmune hepatitis versus drug-induced liver injury[J]. Hepatology, 2011, 54( 3): 931- 939. DOI: 10.1002/hep.24481. [31] HONG-CURTIS J, YEH MMC, JAIN D, et al. Rapid progression of autoimmune hepatitis in the background of primary sclerosing cholangitis[J]. J Clin Gastroenterol, 2004, 38( 10): 906- 909. DOI: 10.1097/00004836-200411000-00015. -

 本文二维码
本文二维码
计量
- 文章访问数: 2511
- HTML全文浏览量: 1076
- PDF下载量: 515
- 被引次数: 0

