
 PDF下载 ( 4200 KB)
PDF下载 ( 4200 KB)

Guideline on the management of cholestasis liver diseases(2021)
-
摘要: 2015年中华医学会肝病学分会和中华医学会消化病学分会制定了我国第一个胆汁淤积性肝病的专家共识。近年来胆汁淤积性肝病的临床研究提供了新的研究数据和资料。为此, 中华医学会肝病学分会自身免疫性肝病学组组织专家组对近年来的文献证据进行了评估, 制定了本指南。本指南共有胆汁淤积性肝病临床诊治推荐意见22条。本指南的目的是为临床胆汁淤积性肝病诊治提供参考和指导。Abstract: In 2015, the Chinese Society of Hepatology and Chinese Society of Gastroenterology issued the consensus on the diagnosis and management of cholestatic liver diseases. In the past years, more data have emerged from clinical practice. Herein, the Autoimmune Liver Disease Group of the Chinese Society of Hepatology organized an expert group to review the evidence and updated the recommendations to formulate the guidelines. There are 22 recommendations on clinical practice of cholestatic liver diseases. The guidelines aim to provide a working reference for the management of cholestatic liver diseases.
-
Key words:
- Cholestatic Liver Disease /
- Diagnosis /
- Therapeutics /
- Guideline
-
表 1 推荐意见的证据等级和推荐强度等级
级别 详细说明 证据质量 高(A) 进一步研究不可能改变对该疗效评估结果的可信度 中(B) 进一步研究有可能影响该疗效评估结果的可信度, 且可能改变该评估结果 低或非常低(C) 进一步研究很有可能影响该疗效评估结果的可信度, 且很可能改变该评估结果 推荐强度 强(1) 明确显示干预措施利大于弊或弊大于利 弱(2) 利弊不确定或无论质量高低的证据均显示利弊相当  下载: 导出CSV
下载: 导出CSV
表 2 胆汁淤积性疾病累及基因
胆汁淤积性疾病 累及基因 Alagille综合征 JAG1, NOTCH2 α1抗胰蛋白酶缺乏症 SERPINA1 α-甲酰基-辅酶A消旋酶缺乏症 AMACR 关节挛缩-肾功能不全- 胆汁淤积综合征 VIPAS39, VPS33B 常染色体隐性多囊肾病 PKHD1 胆汁酸螯合障碍 SLC27A5 胆汁酸再吸收障碍 SLC10A1, SLC10A2 胆汁酸受体缺乏 GPBAR1 胆汁酸合成障碍 CYP7A1 胆道闭锁 SLC51B 脑腱性黄瘤症 CYP27A1 胆固醇酯储积病 LIPA 瓜氨酸血症 SLC25A13 先天性胆汁酸合成障碍 ACOX2, AKR1D1, AMACR, CYP7B1, HSD3B7 囊性纤维化 CFTR D-双功能蛋白缺乏 HSD17B Dubin-Johnson综合征 ABCC2 肝外胆汁淤积症 SLC51B 家族性高胆烷血症 BAAT, TJP2 Lucey-Driscoll综合征 UGT1A1 Crigler-Najjar综合征 UGT1A1 肾小管性范可尼综合征3型 EHHADH 胆囊疾病 ABCB4, ABCG8 遗传性果糖不耐症 ALDOB 鱼鳞病、白细胞空泡、脱发和硬化性胆管炎 CLDN1 妊娠期肝内胆汁淤积症 ABCB4, ATP8B1 Joubert综合征 CC2D2A, MKS1, TMEM216, NPHP1 脂质贮存障碍 SCP2 一过性婴儿肝衰竭 TRMU Meckel综合征 CC2D2A, MKS1, NPHP3, TMEM216 线粒体DNA缺失综合征 DGUOK, POLG, MPV17 新生儿硬化性胆管炎 DCDC2 肾消耗病 INVS, NPHP1, NPHP3, NPHP4 Niemann-Pick病 NPC1, NPC2, SMPD1 北美印第安儿童肝硬化 UTP4 过氧化小体病 PEX1, PEX10, PEX11B, PEX12, PEX13, PEX14, PEX16, PEX19, PEX2, PEX26, PEX3, PEX5, PEX6, PEX7 家族性肝内胆汁淤积症 ABCB11, ABCB4, SLC51A, TJP2, ATP8B1, NR1H4, MYO5B 肾囊肿-糖尿病综合征 HNF1B 肾-肝-胰发育不良1型 PHP3 谷固醇血症 ABCG5, ABCG8 Smith-Le mli-Opitz综合征 DHCR7 醛糖转移酶缺乏 TALDO1 1型酪氨酸血症 FAH
下载: 导出CSV
-
[1] KUNTZ K, KUNTZ HD. Cholestasi hepatology: Principles and practice (2nd edition)[M]. Heidelberg, Germany: Springer Medizin Verlag Heidelberg, 2006: 227-242. [2] WU H, CHEN C, ZIANI S, et al. Fibrotic events in the progression of cholestatic liver disease[J]. Cells, 2021, 10(5): 1107. DOI: 10.3390/cells10051107. [3] Chinese Society of Hepatology, Chinese Medical Association; Chinese Society of Gastroenterology, Chinese Medical Association; Chinese Society of Infectious Diseases, Chinese Medical Association. Consensus on the diagnosis and treatment of cholestasis liver diseases (2015) [J]. J Clin Hepatol, 2015, 31(12): 1989-1999. DOI: 10.3969/j.issn.1001-5256.2015.12.005.中华医学会肝病学分会, 中华医学会消化病学分会, 中华医学会感染病学分会. 胆汁淤积性肝病诊断和治疗共识(2015) [J]. 临床肝胆病杂志, 2015, 31(12): 1989-1999. DOI: 10.3969/j.issn.1001-5256.2015.12.005. [4] SHERLOCK S, DOOLEY J. Cholestasis[M]//SHERLOCK S, DOOLEY J. Disease of the liver and biliary system. 11th ed. Oxford, England: Blackwell Publishing, 2002: 219-240. [5] European Association for the Study of the Liver. EASL clinical practice guidelines: Management of cholestatic liver diseases[J]. J Hepatol, 2009, 51(2): 237-267. DOI: 10.1016/j.jhep.2009.04.009. [6] BURT A, PORTMANN B, FERRELL L. MacSween's pathology of the liver (6th edition)[M]. London, England: Churchill Livingstone Elsevier, 2012: 503-562. [7] LEFKOWITCH JH. Scheuer's liver biopsy interpretation (9th edition)[M]. New York: Elsevier, 2016: 53. [8] HILSCHER MB, KAMATH PS, EATON JE. Cholestatic liver diseases: A primer for generalists and subspecialists[J]. Mayo Clin Proc, 2020, 95(10): 2263-2279. DOI: 10.1016/j.mayocp.2020.01.015. [9] BORTOLINI M, ALMASIO P, BRAY G, et al. Multicentre survey of the prevalence of intrahepatic cholestasis in 2520 consecutive patients with newly diagnosed chronic liver disease[J]. Drug Investigation, 1992, 4(Suppl 4): 83-89. [10] XIE W, CAO Y, XU M, et al. Prognostic significance of elevated cholestatic enzymes for fibrosis and hepatocellular carcinoma in hospital discharged chronic viral hepatitis patients[J]. Sci Rep, 2017, 7(1): 10289. DOI: 10.1038/s41598-017-11111-5. [11] CAO XX, GAO YQ, ZHANG WH, et al. Cholestasis morbidity rate in first-hospitalized patients with chronic liver disease in Shanghai[J]. Chin J Hepatol, 2015, 23(8): 569-573. DOI: 10.3760/cma.j.issn.1007-3418.2015.08.003.曹旬旬, 高月求, 张文宏, 等. 基于上海市住院慢性肝病患者胆汁淤积患病率的调查研究[J]. 中华肝脏病杂志, 2015, 23(8): 569-573. DOI: 10.3760/cma.j.issn.1007-3418.2015.08.003. [12] SIDDIQUE A, KOWDLEY KV. Approach to a patient with elevated serum alkaline phosphatase[J]. Clin Liver Dis, 2012, 16(2): 199-229. DOI: 10.1016/j.cld.2012.03.012. [13] REICHERT MC, HALL RA, KRAWCZYK M, et al. Genetic determinants of cholangiopathies: Molecular and systems genetics[J]. Biochim Biophys Acta Mol Basis Dis, 2018, 1864(4 Pt B): 1484-1490. DOI: 10.1016/j.bbadis.2017.07.029. [14] VITALE G, GITTO S, VUKOTIC R, et al. Familial intrahepatic cholestasis: New and wide perspectives[J]. Dig Liver Dis, 2019, 51(7): 922-933. DOI: 10.1016/j.dld.2019.04.013. [15] PIETERS A, GIJBELS E, COGLIATI B, et al. Biomarkers of cholestasis[J]. Biomark Med, 2021, 15(6): 437-454. DOI: 10.2217/bmm-2020-0691. [16] GELLER S, PETOVIC LM. Evaluation of cholestasis//Biopsy interpretation of the liver (2nd edition)[M]. Philadelphia, USA: Lippincott Williams & Wilkins, 2009: 404-416. [17] PAUMGARTNER G, BEUERS U. Ursodeoxycholic acid in cholestatic liver disease: Mechanisms of action and therapeutic use revisited[J]. Hepatology, 2002, 36(3): 525-531. DOI: 10.1053/jhep.2002.36088. [18] APPANNA G, KALLIS Y. An update on the management of cholestatic liver diseases[J]. Clin Med (Lond), 2020, 20(5): 513-516. DOI: 10.7861/clinmed.2020-0697. [19] WAGNER M, FICKERT P. Drug therapies for chronic cholestatic liver diseases[J]. Annu Rev Pharmacol Toxicol, 2020, 60: 503-527. DOI: 10.1146/annurev-pharmtox-010818-021059. [20] FABRIS L, FIOROTTO R, SPIRLI C, et al. Pathobiology of inherited biliary diseases: A roadmap to understand acquired liver diseases[J]. Nat Rev Gastroenterol Hepatol, 2019, 16(8): 497-511. DOI: 10.1038/s41575-019-0156-4. [21] STAUFER K. Current treatment options for cystic fibrosis-related liver disease[J]. Int J Mol Sci, 2020, 21(22): 8586. DOI: 10.3390/ijms21228586. [22] BOЁLLE PY, DEBRAY D, GUILLOT L, et al. Cystic fibrosis liver disease: Outcomes and risk factors in a large cohort of French patients[J]. Hepatology, 2019, 69(4): 1648-1656. DOI: 10.1002/hep.30148. [23] LI LT, LI ZD, YANG Y, et al. ABCB11 deficiency presenting as transient neonatal cholestasis: Correlation with genotypes and BSEP expression[J]. Liver Int, 2020, 40(11): 2788-2796. DOI: 10.1111/liv.14642. [24] LI L, DEHERAGODA M, LU Y, et al. Hypothyroidism associated with ATP8B1 deficiency[J]. J Pediatr, 2015, 167(6): 1334-1339. e1. DOI: 10.1016/j.jpeds.2015.08.037. [25] LI LT, LI ZD, YANG Y, et al. ABCB11 deficiency presenting as transient neonatal cholestasis: Correlation with genotypes and BSEP expression[J]. Liver Int, 2020, 40(11): 2788-2796. DOI: 10.1111/liv.14642. [26] van WESSEL D, THOMPSON RJ, GONZALES E, et al. Genotype correlates with the natural history of severe bile salt export pump deficiency[J]. J Hepatol, 2020, 73(1): 84-93. DOI: 10.1016/j.jhep.2020.02.007. [27] ZHANG J, LIU LL, GONG JY, et al. TJP2 hepatobiliary disorders: Novel variants and clinical diversity[J]. Hum Mutat, 2020, 41(2): 502-511. DOI: 10.1002/humu.23947. [28] QIU YL, GONG JY, FENG JY, et al. Defects in myosin VB are associated with a spectrum of previously undiagnosed low γ-glutamyltransferase cholestasis[J]. Hepatology, 2017, 65(5): 1655-1669. DOI: 10.1002/hep.29020. [29] ZHANG J, YANG Y, GONG JY, et al. Low-GGT intrahepatic cholestasis associated with biallelic USP53 variants: Clinical, histological and ultrastructural characterization[J]. Liver Int, 2020, 40(5): 1142-1150. DOI: 10.1111/liv.14422. [30] PAN Q, LUO G, QU J, et al. A homozygous R148W mutation in Semaphorin 7A causes progressive familial intrahepatic cholestasis[J]. EMBO Mol Med, 2021, 13(11): e14563. DOI: 10.15252/emmm.202114563. [31] GONZALES E, HARDIKAR W, STORMON M, et al. Efficacy and safety of maralixibat treatment in patients with Alagille syndrome and cholestatic pruritus (ICONIC): A randomised phase 2 study[J]. Lancet, 2021, 398(10311): 1581-1592. DOI: 10.1016/S0140-6736(21)01256-3. [32] EMERICK KM, RAND EB, GOLDMUNTZ E, et al. Features of Alagille syndrome in 92 patients: Frequency and relation to prognosis[J]. Hepatology, 1999, 29(3): 822-829. DOI: 10.1002/hep.510290331. [33] SHNEIDER BL, SPINO C, KAMATH BM, et al. Placebo-controlled randomized trial of an intestinal bile salt transport inhibitor for pruritus in Alagille syndrome[J]. Hepatol Commun, 2018, 2(10): 1184-1198. DOI: 10.1002/hep4.1244. [34] FLOREANI A, GERVASI MT. New insights on intrahepatic cholestasis of pregnancy[J]. Clin Liver Dis, 2016, 20(1): 177-189. DOI: 10.1016/j.cld.2015.08.010. [35] WOOD AM, LIVINGSTON EG, HUGHES BL, et al. Intrahepatic cholestasis of pregnancy: A review of diagnosis and management[J]. Obstet Gynecol Surv, 2018, 73(2): 103-109. DOI: 10.1097/OGX.0000000000000524. [36] SMITH DD, ROOD KM. Intrahepatic cholestasis of pregnancy[J]. Clin Obstet Gynecol, 2020, 63(1): 134-151. DOI: 10.1097/GRF.0000000000000495. [37] LANGEDIJK J, BEUERS UH, OUDE ELFERINK R. Cholestasis-associated pruritus and its pruritogens[J]. Front Med (Lausanne), 2021, 8: 639674. DOI: 10.3389/fmed.2021.639674. [38] PATEL SP, VASAVDA C, HO B, et al. Cholestatic pruritus: Emerging mechanisms and therapeutics[J]. J Am Acad Dermatol, 2019, 81(6): 1371-1378. DOI: 10.1016/j.jaad.2019.04.035. [39] BERGASA NV, JONES EA. Assessment of the visual analogue score in the evaluation of the pruritus of cholestasis[J]. J Clin Transl Hepatol, 2017, 5(3): 203-207. DOI: 10.14218/JCTH.2017.00001. [40] THÉBAUT A, HABES D, GOTTRAND F, et al. Sertraline as an additional treatment for cholestatic pruritus in children[J]. J Pediatr Gastroenterol Nutr, 2017, 64(3): 431-435. DOI: 10.1097/MPG.0000000000001385. [41] WEBB GJ, RAHMAN SR, LEVY C, et al. Low risk of hepatotoxicity from rifampicin when used for cholestatic pruritus: A cross-disease cohort study[J]. Aliment Pharmacol Ther, 2018, 47(8): 1213-1219. DOI: 10.1111/apt.14579. [42] PHAW NA, LEIGHTON J, DYSON JK, et al. Managing cognitive symptoms and fatigue in cholestatic liver disease[J]. Expert Rev Gastroenterol Hepatol, 2021, 15(3): 235-241. DOI: 10.1080/17474124.2021.1844565. [43] SWAIN MG, JONES D. Fatigue in chronic liver disease: New insights and therapeutic approaches[J]. Liver Int, 2019, 39(1): 6-19. DOI: 10.1111/liv.13919. [44] DUBROVSKY A, BOWLUS CL. Statins, fibrates, and other peroxisome proliferator-activated receptor agonists for the treatment of cholestatic liver diseases[J]. Gastroenterol Hepatol (N Y), 2020, 16(1): 31-38. [45] European Association for the Study of the Liver. EASL clinical practice guidelines on nutrition in chronic liver disease[J]. J Hepatol, 2019, 70(1): 172-193. DOI: 10.1016/j.jhep.2018.06.024. -


 本文二维码
本文二维码
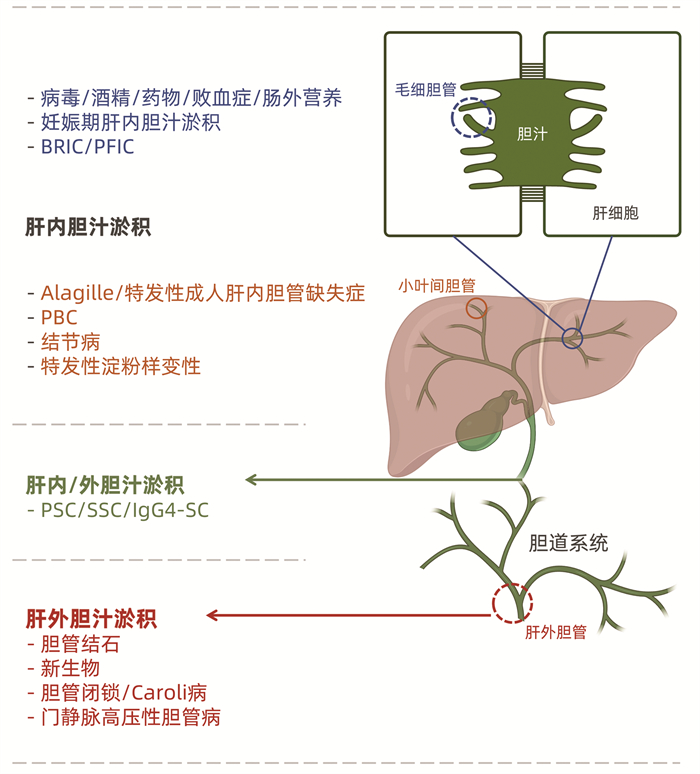
图(2) / 表(2)
计量
- 文章访问数: 5780
- HTML全文浏览量: 3671
- PDF下载量: 1983
- 被引次数: 0

