
 PDF下载 ( 1051 KB)
PDF下载 ( 1051 KB)

孟德尔随机化在胰腺癌研究中的应用现状与展望
DOI: 10.12449/JCH241033
利益冲突声明:本文不存在任何利益冲突。
作者贡献声明:杜凯豪负责课题设计,论文的撰写与修改;罗兰明慧、东小鸽、蒋威参与文献搜集,修改论文;王展、侯立朝负责拟定写作思路,指导撰写文章,并最后定稿。
Current status and prospects of the application of Mendelian randomization in pancreatic cancer research
-
摘要: 胰腺癌的发病隐匿性强,治疗困难,早期诊断和治疗存在诸多局限性。本文总结了孟德尔随机化(MR)在胰腺癌风险因素探索中的应用进展,特别分析了肠道微生态、生活方式、代谢性疾病等因素的因果关系。通过大规模基因组关联研究(GWAS)数据,MR分析揭示了若干与胰腺癌风险相关的生物标志物。双样本MR是当前研究中的常用方法,包括逆方差加权法、加权中位数、MR-Egger法等,有助于从遗传角度解释疾病的因果网络。尽管MR策略可以为理解胰腺癌的病因学提供新视角,但其在数据合成、工具变量选择及多效性评估方面仍需谨慎。新兴分析模型如贝叶斯加权MR(BWMR)、CAUSE和多变量MR(MVMR)等,为综合评估多重风险因素及其相互关系带来新的可能。未来,结合上述方法及累积增多的遗传流行病学资料,MR分析预期能够为发现胰腺癌的潜在治疗靶点和制订预防策略提供更为坚实的证据基础。Abstract: Pancreatic cancer often has an insidious onset and difficulties in treatment, with various limitations in early diagnosis and treatment. This article reviews the application of Mendelian randomization (MR) in exploring the risk factors for pancreatic cancer, with a special focus on the causal relationships of factors such as gut microbiota, lifestyle, and metabolic diseases. Leveraging data from large-scale genome-wide association studies (GWAS), MR analysis has revealed several biomarkers associated with the risk of pancreatic cancer. The two-sample MR approach is commonly used in current research, including the methods such as Inverse Variance Weighted, Weighted Median, and MR-Egger, which helps to explain the causal network of the disease from a genetic perspective. While MR strategy provides a new perspective for understanding the etiology of pancreatic cancer, caution is still needed in data synthesis, selection of instrumental variables, and pleiotropy assessment. The use of emerging analytical models such as BWMR, CAUSE, and MVMR offers new possibilities for the comprehensive evaluation of multiple risk factors and their interaction. In the future, with the combination of these methods and the ever-increasing genetic epidemiological data, MR analysis is expected to provide more solid evidence for identifying potential therapeutic targets for pancreatic cancer and formulating prevention strategies.
-
Key words:
- Pancreatic Neoplasms /
- Mendelian Randomization Analysis /
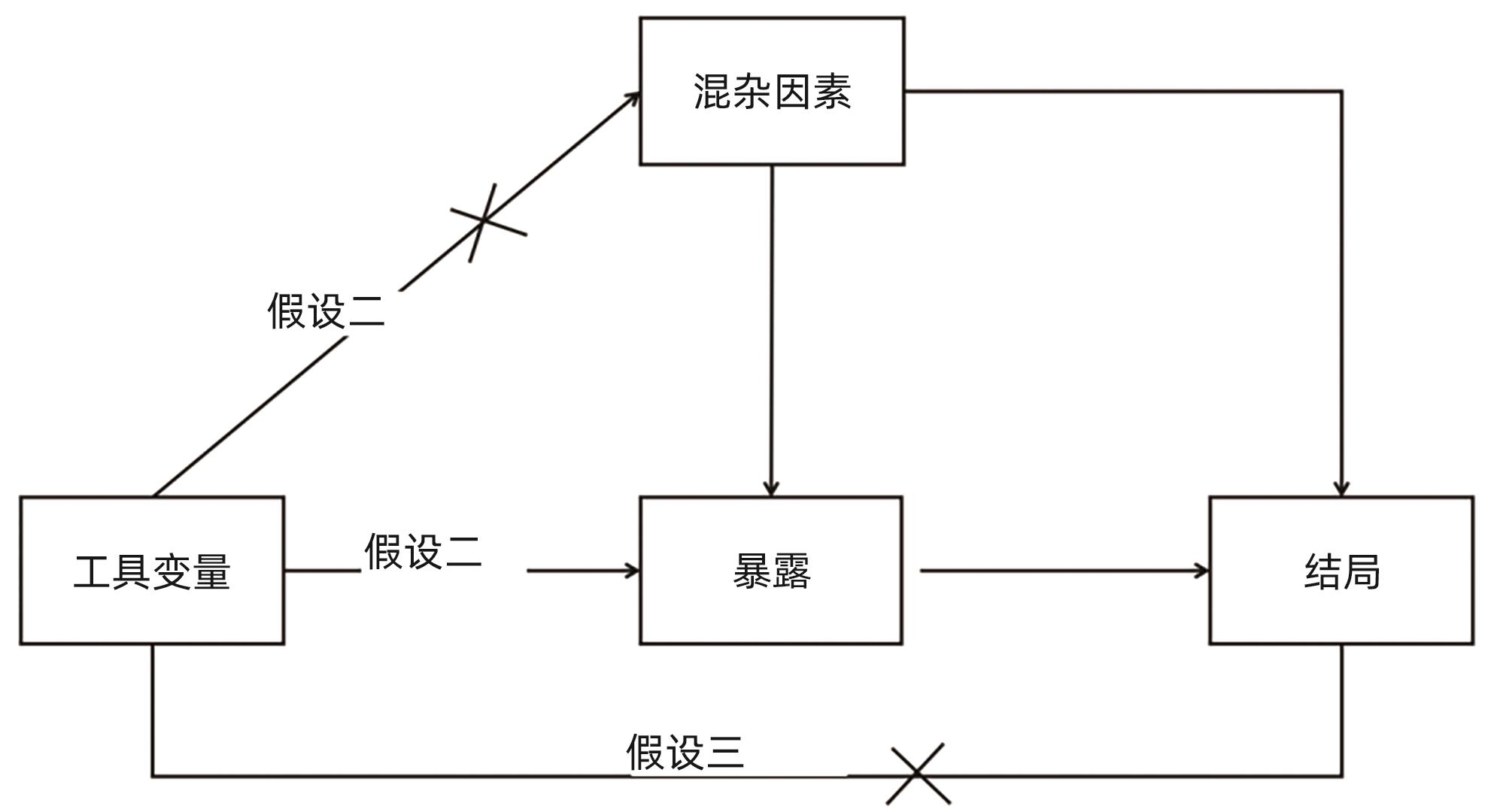
- Causality
-
表 1 肠道菌群MR
Table 1. Gut microbiota MR
GM OR 95%CI P值 研究1[25] Senegalimassilia 0.635 0.406~0.998 0.049 Odoribacter 1.899 1.157~3.116 0.011 Ruminiclostridium 9 1.976 1.128~3.461 0.017 Ruminococcaceae(UCG011) 1.433 1.072~1.916 0.015 Streptococcus 1.712 1.071~1.736 0.025 研究2[26] Lentisphaerae 0.726 0.553~0.953 0.021 Lentisphaeria 0.697 0.521~0.932 0.015 Victivallales 0.697 0.521~0.932 0.015 Lachnospiraceae UCG004 0.655 0.434~0.988 0.043 Erysipelotrichia 1.616 1.025~2.547 0.039 Erysipelotrichales 1.616 1.025~2.547 0.039 Erysipelotrichaceae 1.616 1.025~2.547 0.039 Flavonifractor 1.857 1.120~3.079 0.016 Terrisporobacter 1.671 1.061~2.632 0.027  下载: 导出CSV
下载: 导出CSV
表 3 炎症、免疫因素MR
Table 3. Inflammatory and immune factors MR
暴露 结局 方法 OR 95%CI P值 溃疡性结肠炎 胰腺癌 IVW 0.946 0.830~1.079 0.409 WM 0.910 0.753~1.101 0.332 MR-Egger 0.951 0.648~1.397 0.799 克罗恩病 胰腺癌 IVW 1.111 1.015~1.213 0.022 WM 1.120 0.970~1.292 0.119 MR-Egger 1.338 1.064~1.683 0.015 IL-1RA 急性胰腺炎 IVW 0.87 0.77~0.97 0.003 慢性胰腺炎 0.73 0.65~0.82 2.93×10-8 胰腺癌 0.86 0.77~0.96 0.009
下载: 导出CSV
-
[1] ARNOLD M, ABNET CC, NEALE RE, et al. Global burden of 5 major types of gastrointestinal cancer[J]. Gastroenterology, 2020, 159( 1): 335- 349. e 15. DOI: 10.1053/j.gastro.2020.02.068. [2] ILIC M, ILIC I. Epidemiology of pancreatic cancer[J]. World J Gastroenterol, 2016, 22( 44): 9694- 9705. DOI: 10.3748/wjg.v22.i44.9694. [3] SUNG H, FERLAY J, SIEGEL RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries[J]. CA Cancer J Clin, 2021, 71( 3): 209- 249. DOI: 10.3322/caac.21660. [4] FERLAY J, SOERJOMATARAM I, DIKSHIT R, et al. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012[J]. Int J Cancer, 2015, 136( 5): E359- E386. DOI: 10.1002/ijc.29210. [5] HIDALGO M, CASCINU S, KLEEFF J, et al. Addressing the challenges of pancreatic cancer: Future directions for improving outcomes[J]. Pancreatology, 2015, 15( 1): 8- 18. DOI: 10.1016/j.pan.2014.10.001. [6] General Office of National Health Commission. Standard for diagnosis and treatment of pancreatic cancer(2022 edition)[J]. J Clin Hepatol, 2022, 38( 5): 1006- 1015. DOI: 10.3969/j.issn.1001-5256.2022.05.007.国家卫生健康委办公厅. 胰腺癌诊疗指南(2022年版)[J]. 临床肝胆病杂志, 2022, 38( 5): 1006- 1015. DOI: 10.3969/j.issn.1001-5256.2022.05.007. [7] MILLIKAN KW, DEZIEL DJ, SILVERSTEIN JC, et al. Prognostic factors associated with resectable adenocarcinoma of the head of the pancreas[J]. Am Surg, 1999, 65( 7): 618-623; discussion 623-624. [8] KOLBEINSSON HM, CHANDANA S, WRIGHT GP, et al. Pancreatic cancer: A review of current treatment and novel therapies[J]. J Invest Surg, 2023, 36( 1): 2129884. DOI: 10.1080/08941939.2022.2129884. [9] PARKIN DM, BOYD L, WALKER LC. 16. The fraction of cancer attributable to lifestyle and environmental factors in the UK in 2010[J]. Br J Cancer, 2011, 105( Suppl 2): S77- S81. DOI: 10.1038/bjc.2011.489. [10] WILLETT WC. Diet and cancer[J]. Oncol, 2000, 5( 5): 393- 404. DOI: 10.1634/theoncologist.5-5-393. [11] BOSETTI C, BERTUCCIO P, NEGRI E, et al. Pancreatic cancer: Overview of descriptive epidemiology[J]. Mol Carcinog, 2012, 51( 1): 3- 13. DOI: 10.1002/mc.20785. [12] ANAND P, KUNNUMAKKARA AB, SUNDARAM C, et al. Cancer is a preventable disease that requires major lifestyle changes[J]. Pharm Res, 2008, 25( 9): 2097- 2116. DOI: 10.1007/s11095-008-9661-9. [13] WANG LN, ZHANG ZF. Mendelian randomization approach, used for causal inferences[J]. Chin J Epidemiol, 2017, 38( 4): 547- 552. DOI: 10.3760/cma.j.issn.0254-6450.2017.04.027.王莉娜, ZHANG ZuoFeng. 孟德尔随机化法在因果推断中的应用[J]. 中华流行病学杂志, 2017, 38( 4): 547- 552. DOI: 10.3760/cma.j.issn.0254-6450.2017.04.027. [14] DAVEY SMITH G, HEMANI G. Mendelian randomization: Genetic anchors for causal inference in epidemiological studies[J]. Hum Mol Genet, 2014, 23( R1): R89- R98. DOI: 10.1093/hmg/ddu328. [15] ZHENG J, BAIRD D, BORGES MC, et al. Recent developments in Mendelian randomization studies[J]. Curr Epidemiol Rep, 2017, 4( 4): 330- 345. DOI: 10.1007/s40471-017-0128-6. [16] LAWLOR DA, HARBORD RM, STERNE JA, et al. Mendelian randomization: Using genes as instruments for making causal inferences in epidemiology[J]. Stat Med, 2008, 27( 8): 1133- 1163. DOI: 10.1002/sim.3034. [17] VERBANCK M, CHEN CY, NEALE B, et al. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases[J]. Nat Genet, 2018, 50( 5): 693- 698. DOI: 10.1038/s41588-018-0099-7. [18] BOWDEN J, DAVEY SMITH G, BURGESS S. Mendelian randomization with invalid instruments: Effect estimation and bias detection through Egger regression[J]. Int J Epidemiol, 2015, 44( 2): 512- 525. DOI: 10.1093/ije/dyv080. [19] ZHAO J, MING JS, HU XH, et al. Bayesian weighted Mendelian randomization for causal inference based on summary statistics[J]. Bioinformatics, 2020, 36( 5): 1501- 1508. DOI: 10.1093/bioinformatics/btz749. [20] LAWLOR DA. Commentary: Two-sample Mendelian randomization: Opportunities and challenges[J]. Int J Epidemiol, 2016, 45( 3): 908- 915. DOI: 10.1093/ije/dyw127. [21] BURGESS S, BUTTERWORTH A, THOMPSON SG. Mendelian randomization analysis with multiple genetic variants using summarized data[J]. Genet Epidemiol, 2013, 37( 7): 658- 665. DOI: 10.1002/gepi.21758. [22] YU TQ, XU WT, SU YN, et al. Basic principles, methods and limitations of Mendelian randomization studies[J]. Chin J Evid-Based Med, 2021, 21( 10): 1227- 1234. DOI: 10.7507/1672-2531.202107008.于天琦, 徐文涛, 苏雅娜, 等. 孟德尔随机化研究基本原理、方法和局限性[J]. 中国循证医学杂志, 2021, 21( 10): 1227- 1234. DOI: 10.7507/1672-2531.202107008. [23] BOWDEN J, DAVEY SMITH G, HAYCOCK PC, et al. Consistent estimation in Mendelian randomization with some invalid instruments using a weighted Median estimator[J]. Genet Epidemiol, 2016, 40( 4): 304- 314. DOI: 10.1002/gepi.21965. [24] BOWDEN J, DEL GRECO MF, MINELLI C, et al. Assessing the suitability of summary data for two-sample Mendelian randomization analyses using MR-Egger regression: The role of the I2 statistic[J]. Int J Epidemiol, 2016, 45( 6): 1961- 1974. DOI: 10.1093/ije/dyw220. [25] JIANG ZC, MOU YP, WANG HJ, et al. Causal effect between gut microbiota and pancreatic cancer: A two-sample Mendelian randomization study[J]. BMC Cancer, 2023, 23( 1): 1091. DOI: 10.1186/s12885-023-11493-y. [26] ZHU SJ, DING Z. Association between gut microbiota and seven gastrointestinal diseases: A Mendelian randomized study[J]. J Gene Med, 2024, 26( 1): e3623. DOI: 10.1002/jgm.3623. [27] ZHANG XN, ZHAO H, MAN JY, et al. Investigating causal associations of diet-derived circulating antioxidants with the risk of digestive system cancers: A Mendelian randomization study[J]. Nutrients, 2022, 14( 15): 3237. DOI: 10.3390/nu14153237. [28] MIN Y, LIU ZR, LI RD, et al. Association between inflammatory bowel disease and pancreatic cancer: Results from the two-sample Mendelian randomization study[J]. Front Oncol, 2023, 13: 1155123. DOI: 10.3389/fonc.2023.1155123. [29] PIERCE BL, AHSAN H, VANDERWEELE TJ. Power and instrument strength requirements for Mendelian randomization studies using multiple genetic variants[J]. Int J Epidemiol, 2011, 40( 3): 740- 752. DOI: 10.1093/ije/dyq151. [30] ADAK A, KHAN MR. An insight into gut microbiota and its functionalities[J]. Cell Mol Life Sci, 2019, 76( 3): 473- 493. DOI: 10.1007/s00018-018-2943-4. [31] de VOS WM, TILG H, van HUL M, et al. Gut microbiome and health: Mechanistic insights[J]. Gut, 2022, 71( 5): 1020- 1032. DOI: 10.1136/gutjnl-2021-326789. [32] LIU MY. Arid1a: A gatekeeper in the development of pancreatic cancer from a rare precursor lesion[J]. Gastroenterology, 2022, 163( 2): 371- 373. DOI: 10.1053/j.gastro.2022.05.046. [33] CAI J, CHEN HD, LU M, et al. Advances in the epidemiology of pancreatic cancer: Trends, risk factors, screening, and prognosis[J]. Cancer Lett, 2021, 520: 1- 11. DOI: 10.1016/j.canlet.2021.06.027. [34] TONG Y, GAO HR, QI QC, et al. High fat diet, gut microbiome and gastrointestinal cancer[J]. Theranostics, 2021, 11( 12): 5889- 5910. DOI: 10.7150/thno.56157. [35] COHEN LJ, CHO JH, GEVERS D, et al. Genetic factors and the intestinal microbiome guide development of microbe-based therapies for inflammatory bowel diseases[J]. Gastroenterology, 2019, 156( 8): 2174- 2189. DOI: 10.1053/j.gastro.2019.03.017. [36] WONG SH, YU J. Gut microbiota in colorectal cancer: Mechanisms of action and clinical applications[J]. Nat Rev Gastroenterol Hepatol, 2019, 16( 11): 690- 704. DOI: 10.1038/s41575-019-0209-8. [37] MA C, HAN MJ, HEINRICH B, et al. Gut microbiome-mediated bile acid metabolism regulates liver cancer via NKT cells[J]. Science, 2018, 360( 6391): eaan5931. DOI: 10.1126/science.aan5931. [38] ERTZ-ARCHAMBAULT N, KEIM P, von HOFF D. Microbiome and pancreatic cancer: A comprehensive topic review of literature[J]. World J Gastroenterol, 2017, 23( 10): 1899- 1908. DOI: 10.3748/wjg.v23.i10.1899. [39] YANG QY, ZHANG JH, ZHU Y. Potential roles of the gut microbiota in pancreatic carcinogenesis and therapeutics[J]. Front Cell Infect Microbiol, 2022, 12: 872019. DOI: 10.3389/fcimb.2022.872019. [40] PUSHALKAR S, HUNDEYIN M, DALEY D, et al. The pancreatic cancer microbiome promotes oncogenesis by induction of innate and adaptive immune suppression[J]. Cancer Discov, 2018, 8( 4): 403- 416. DOI: 10.1158/2159-8290.CD-17-1134. [41] SETHI V, KURTOM S, TARIQUE M, et al. Gut microbiota promotes tumor growth in mice by modulating immune response[J]. Gastroenterology, 2018, 155( 1): 33- 37. e 6. DOI: 10.1053/j.gastro.2018.04.001. [42] YU Q, NEWSOME RC, BEVERIDGE M, et al. Intestinal microbiota modulates pancreatic carcinogenesis through intratumoral natural killer cells[J]. Gut Microbes, 2022, 14( 1): 2112881. DOI: 10.1080/19490976.2022.2112881. [43] CARINI F, MAZZOLA M, RAPPA F, et al. Colorectal carcinogenesis: Role of oxidative stress and antioxidants[J]. Anticancer Res, 2017, 37( 9): 4759- 4766. DOI: 10.21873/anticanres.11882. [44] SHIMIZU Y, TAMURA T, KEMMOCHI A, et al. Oxidative stress and Liver X Receptor agonist induce hepatocellular carcinoma in Non-alcoholic steatohepatitis model[J]. J Gastroenterol Hepatol, 2021, 36( 3): 800- 810. DOI: 10.1111/jgh.15239. [45] AKBARI A, MAJD HM, RAHNAMA R, et al. Cross-talk between oxidative stress signaling and microRNA regulatory systems in carcinogenesis: Focused on gastrointestinal cancers[J]. Biomedecine Pharmacother, 2020, 131: 110729. DOI: 10.1016/j.biopha.2020.110729. [46] KLAUNIG JE. Oxidative stress and cancer[J]. Curr Pharm Des, 2018, 24( 40): 4771- 4778. DOI: 10.2174/1381612825666190215121712. [47] YIN LL, YAN HH, CHEN KD, et al. Diet-derived circulating antioxidants and risk of digestive system tumors: A Mendelian randomization study[J]. Nutrients, 2022, 14( 16): 3274. DOI: 10.3390/nu14163274. [48] SEMPERE-RUBIO N, AGUAS M, FAUBEL R. Association between chronotype, physical activity and sedentary behaviour: A systematic review[J]. Int J Environ Res Public Health, 2022, 19( 15): 9646. DOI: 10.3390/ijerph19159646. [49] MAZRI FH, MANAF ZA, SHAHAR S, et al. The association between chronotype and dietary pattern among adults: A scoping review[J]. Int J Environ Res Public Health, 2019, 17( 1): 68. DOI: 10.3390/ijerph17010068. [50] SROUR B, PLANCOULAINE S, ANDREEVA VA, et al. Circadian nutritional behaviours and cancer risk: New insights from the NutriNet-santé prospective cohort study: Disclaimers[J]. Int J Cancer, 2018, 143( 10): 2369- 2379. DOI: 10.1002/ijc.31584. [51] CARASSO S, FISHMAN B, LASK LS, et al. Metagenomic analysis reveals the signature of gut microbiota associated with human chronotypes[J]. FASEB J, 2021, 35( 11): e22011. DOI: 10.1096/fj.202100857RR. [52] MENG CT, BAI CM, BROWN TD, et al. Human gut microbiota and gastrointestinal cancer[J]. Genomics Proteomics Bioinformatics, 2018, 16( 1): 33- 49. DOI: 10.1016/j.gpb.2017.06.002. [53] FENG Q, LIANG SS, JIA HJ, et al. Gut microbiome development along the colorectal adenoma-carcinoma sequence[J]. Nat Commun, 2015, 6: 6528. DOI: 10.1038/ncomms7528. [54] YUAN S, MASON AM, TITOVA OE, et al. Morning chronotype and digestive tract cancers: Mendelian randomization study[J]. Int J Cancer, 2023, 152( 4): 697- 704. DOI: 10.1002/ijc.34284. [55] RISK FACTOR COLLABORATION NCD-RISC) NCD. Worldwide trends in diabetes since 1980: A pooled analysis of 751 population-based studies with 4.4 million participants[J]. Lancet, 2016, 387( 10027): 1513- 1530. DOI: 10.1016/S0140-6736(16)00618-8. [56] TSILIDIS KK, KASIMIS JC, LOPEZ DS, et al. Type 2 diabetes and cancer: Umbrella review of meta-analyses of observational studies[J]. BMJ, 2015, 350: g7607. DOI: 10.1136/bmj.g7607. [57] PEARSON-STUTTARD J, PAPADIMITRIOU N, MARKOZANNES G, et al. Type 2 diabetes and cancer: An umbrella review of observational and Mendelian randomization studies[J]. Cancer Epidemiol Biomarkers Prev, 2021, 30( 6): 1218- 1228. DOI: 10.1158/1055-9965.EPI-20-1245. [58] SHEN BY, LI YY, SHENG CS, et al. Association between age at diabetes onset or diabetes duration and subsequent risk of pancreatic cancer: Results from a longitudinal cohort and Mendelian randomization study[J]. Lancet Reg Health West Pac, 2023, 30: 100596. DOI: 10.1016/j.lanwpc.2022.100596. [59] INCIO J, LIU H, SUBOJ P, et al. Obesity-induced inflammation and desmoplasia promote pancreatic cancer progression and resistance to chemotherapy[J]. Cancer Discov, 2016, 6( 8): 852- 869. DOI: 10.1158/2159-8290.CD-15-1177. [60] PARK JH, AMERI AH, DEMPSEY KE, et al. Nuclear IL-33/SMAD signaling axis promotes cancer development in chronic inflammation[J]. EMBO J, 2021, 40( 7): e106151. DOI: 10.15252/embj.2020106151. [61] YUDKIN JS, STEHOUWER CD, EMEIS JJ, et al. C-reactive protein in healthy subjects: Associations with obesity, insulin resistance, and endothelial dysfunction: A potential role for cytokines originating from adipose tissue?[J]. Arterioscler Thromb Vasc Biol, 1999, 19( 4): 972- 978. DOI: 10.1161/01.atv.19.4.972. [62] XU HY, BARNES GT, YANG Q, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance[J]. J Clin Invest, 2003, 112( 12): 1821- 1830. DOI: 10.1172/JCI19451. [63] SINGH A, MAYENGBAM SS, YADUVANSHI H, et al. Obesity programs macrophages to support cancer progression[J]. Cancer Res, 2022, 82( 23): 4303- 4312. DOI: 10.1158/0008-5472.CAN-22-1257. [64] XIA B, HE QS, PAN YH, et al. Metabolic syndrome and risk of pancreatic cancer: A population-based prospective cohort study[J]. Int J Cancer, 2020, 147( 12): 3384- 3393. DOI: 10.1002/ijc.33172. [65] LI ZQ, JIN LQ, XIA L, et al. Body mass index, C-reactive protein, and pancreatic cancer: A Mendelian randomization analysis to investigate causal pathways[J]. Front Oncol, 2023, 13: 1042567. DOI: 10.3389/fonc.2023.1042567. [66] SON J, LYSSIOTIS CA, YING HQ, et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway[J]. Nature, 2013, 496( 7443): 101- 105. DOI: 10.1038/nature12040. [67] CHEUNG PK, MA MH, TSE HF, et al. The applications of metabolomics in the molecular diagnostics of cancer[J]. Expert Rev Mol Diagn, 2019, 19( 9): 785- 793. DOI: 10.1080/14737159.2019.1656530. [68] XIE GX, LU LG, QIU YP, et al. Plasma metabolite biomarkers for the detection of pancreatic cancer[J]. J Proteome Res, 2015, 14( 2): 1195- 1202. DOI: 10.1021/pr501135f. [69] LI QX, JIN M, LIU YH, et al. Gut microbiota: Its potential roles in pancreatic cancer[J]. Front Cell Infect Microbiol, 2020, 10: 572492. DOI: 10.3389/fcimb.2020.572492. [70] ZHONG H, LIU S, ZHU JJ, et al. Elucidating the role of blood metabolites on pancreatic cancer risk using two-sample Mendelian randomization analysis[J]. Int J Cancer, 2024, 154( 5): 852- 862. DOI: 10.1002/ijc.34771. [71] NADEEM MS, KUMAR V, AL-ABBASI FA, et al. Risk of colorectal cancer in inflammatory bowel diseases[J]. Semin Cancer Biol, 2020, 64: 51- 60. DOI: 10.1016/j.semcancer.2019.05.001. [72] ZHANG HM, ZHANG MM, CHEN XF, et al. Risk of malignancy in patients with inflammatory bowel disease: A population-based cohort study from China[J]. Int J Cancer, 2022, 150( 11): 1770- 1778. DOI: 10.1002/ijc.33932. [73] JUNG YS, HAN M, PARK S, et al. Cancer risk in the early stages of inflammatory bowel disease in Korean patients: A nationwide population-based study[J]. J Crohns Colitis, 2017, 11( 8): 954- 962. DOI: 10.1093/ecco-jcc/jjx040. [74] ÅH EVERHOV, ERICHSEN R, SACHS MC, et al. Inflammatory bowel disease and pancreatic cancer: A Scandinavian register-based cohort study 1969-2017[J]. Aliment Pharmacol Ther, 2020, 52( 1): 143- 154. DOI: 10.1111/apt.15785. [75] LO B, ZHAO M, VIND I, et al. The risk of extraintestinal cancer in inflammatory bowel disease: A systematic review and meta-analysis of population-based cohort studies[J]. Clin Gastroenterol Hepatol, 2021, 19( 6): 1117- 1138. e 19. DOI: 10.1016/j.cgh.2020.08.015. [76] DENHAM W, YANG J, FINK G, et al. Gene targeting demonstrates additive detrimental effects of interleukin 1 and tumor necrosis factor during pancreatitis[J]. Gastroenterology, 1997, 113( 5): 1741- 1746. DOI: 10.1053/gast.1997.v113.pm9352880. [77] MARRACHE F, TU SP, BHAGAT G, et al. Overexpression of interleukin-1beta in the murine pancreas results in chronic pancreatitis[J]. Gastroenterology, 2008, 135( 4): 1277- 1287. DOI: 10.1053/j.gastro.2008.06.078. [78] TAKAHASHI R, MACCHINI M, SUNAGAWA M, et al. Interleukin-1β- induced pancreatitis promotes pancreatic ductal adenocarcinoma via B lymphocyte-mediated immune suppression[J]. Gut, 2021, 70( 2): 330- 341. DOI: 10.1136/gutjnl-2019-319912. [79] ZHANG Y, CHEN XY, WANG HM, et al. Innate immune mediator, Interleukin-1 receptor accessory protein(IL1RAP), is expressed and pro-tumorigenic in pancreatic cancer[J]. J Hematol Oncol, 2022, 15( 1): 70. DOI: 10.1186/s13045-022-01286-4. [80] TJOMSLAND V, SANDNES D, POMIANOWSKA E, et al. The TGFβ- SMAD3 pathway inhibits IL-1α induced interactions between human pancreatic stellate cells and pancreatic carcinoma cells and restricts cancer cell migration[J]. J Exp Clin Cancer Res, 2016, 35( 1): 122. DOI: 10.1186/s13046-016-0400-5. [81] MAKER AV, KATABI N, QIN LX, et al. Cyst fluid interleukin-1beta(IL1beta) levels predict the risk of carcinoma in intraductal papillary mucinous neoplasms of the pancreas[J]. Clin Cancer Res, 2011, 17( 6): 1502- 1508. DOI: 10.1158/1078-0432.CCR-10-1561. [82] YUAN S, MIAO YY, RUAN XX, et al. Therapeutic role of interleukin-1 receptor antagonist in pancreatic diseases: Mendelian randomization study[J]. Front Immunol, 2023, 14: 1240754. DOI: 10.3389/fimmu.2023.1240754. [83] GRECO M FD, MINELLI C, SHEEHAN NA, et al. Detecting pleiotropy in Mendelian randomisation studies with summary data and a continuous outcome[J]. Stat Med, 2015, 34( 21): 2926- 2940. DOI: 10.1002/sim.6522. [84] WANG X, GAO HY, ZENG YY, et al. A Mendelian analysis of the relationships between immune cells and breast cancer[J]. Front Oncol, 2024, 14: 1341292. DOI: 10.3389/fonc.2024.1341292. [85] MORRISON J, KNOBLAUCH N, MARCUS JH, et al. Mendelian randomization accounting for correlated and uncorrelated pleiotropic effects using genome-wide summary statistics[J]. Nat Genet, 2020, 52( 7): 740- 747. DOI: 10.1038/s41588-020-0631-4. [86] BARANOVA A, CAO HB, ZHANG FQ. Causal effect of COVID-19 on Alzheimer’s disease: A Mendelian randomization study[J]. J Med Virol, 2023, 95( 1): e28107. DOI: 10.1002/jmv.28107. [87] MICHAËLSSON M, YUAN S, MELHUS H, et al. The impact and causal directions for the associations between diagnosis of ADHD, socioeconomic status, and intelligence by use of a bi-directional two-sample Mendelian randomization design[J]. BMC Med, 2022, 20( 1): 106. DOI: 10.1186/s12916-022-02314-3. [88] SANDERSON E. Multivariable Mendelian randomization and mediation[J]. Cold Spring Harb Perspect Med, 2021, 11( 2): a038984. DOI: 10.1101/cshperspect.a038984. [89] WILLER CJ, SCHMIDT EM, SENGUPTA S, et al. Discovery and refinement of loci associated with lipid levels[J]. Nat Genet, 2013, 45( 11): 1274- 1283. DOI: 10.1038/ng.2797. [90] WÜRTZ P, KANGAS AJ, SOININEN P, et al. Lipoprotein subclass profiling reveals pleiotropy in the genetic variants of lipid risk factors for coronary heart disease: A note on Mendelian randomization studies[J]. J Am Coll Cardiol, 2013, 62( 20): 1906- 1908. DOI: 10.1016/j.jacc.2013.07.085. [91] BURGESS S, THOMPSON SG. Multivariable Mendelian randomization: The use of pleiotropic genetic variants to estimate causal effects[J]. Am J Epidemiol, 2015, 181( 4): 251- 260. DOI: 10.1093/aje/kwu283. [92] BURGESS S, DUDBRIDGE F, THOMPSON SG. Re:“Multivariable Mendelian randomization: The use of pleiotropic genetic variants to estimate causal effects”[J]. Am J Epidemiol, 2015, 181( 4): 290- 291. DOI: 10.1093/aje/kwv017. [93] KEMP JP, SAYERS A, SMITH GD, et al. Using Mendelian randomization to investigate a possible causal relationship between adiposity and increased bone mineral density at different skeletal sites in children[J]. Int J Epidemiol, 2016, 45( 5): 1560- 1572. DOI: 10.1093/ije/dyw079. -

 本文二维码
本文二维码
图(2) / 表(3)
计量
- 文章访问数: 1751
- HTML全文浏览量: 871
- PDF下载量: 124
- 被引次数: 0

