
 PDF下载 ( 1943 KB)
PDF下载 ( 1943 KB)

NLRP3基因敲减对高脂高糖饮食诱导的非酒精性脂肪性肝炎小鼠模型的影响
DOI: 10.12449/JCH240514
Effect of NOD‑like receptor family pyrin domain containing 3 knockdown on a mouse model of nonalcoholic steatohepatitis induced by high-fat high-carbohydrate diet
-
摘要:
目的 探讨NOD样受体热蛋白结构域相关蛋白3(NLRP3)基因敲减对高脂高糖诱导的非酒精性脂肪性肝炎(NASH)小鼠模型的影响。 方法 将44只小鼠随机分为正常饮食组(CON)20只,高脂高糖造模组(HFHC)24只。造模14周末,随机选取4只HFHC组小鼠进行腺相关病毒9(AAV9)尾静脉注射预实验,4周后验证NLRP3敲减模型是否成功。18周末确认敲减成功后,对剩余40只小鼠进行AAV9一次性尾静脉注射,分为CON+NLRP3敲减阴性对照组(CON+NLRP3-NC)、CON+NLRP3敲减组(CON+NLRP3-KD)、HFHC+NLRP3-NC及HHFHC+NLRP3-KD组,每组10只,继续造模6周。24周末取材观察炎症小体活化效应,检测小鼠体质量、肝质量、肝指数及糖代谢(空腹血糖、空腹胰岛素及HOMA-IR指数)指标;检测小鼠肝脂质含量(肝组织TG及油红O染色)、肝脏炎症(血清ALT活性、HE染色及炎症相关基因)及肝纤维化(天狼星红染色及纤维化相关基因)指标。计量资料多组间比较使用单因素方差分析,进一步两两比较采用LSD-t检验。 结果 与CON+NLRP3-NC组相比,Western Blot结果提示,HFHC+NLRP3-NC组的NLRP3、pro-Caspase1、Caspase1、ASC及IL-1β蛋白水平均升高,HFHC+NLRP3-KD组均降低(P值均<0.05);HFHC+NLRP3-NC组小鼠体质量、肝质量、肝指数及糖代谢指标均有不同程度升高,HFHC+NLRP3-KD组均显著改善(P值均<0.05);在肝脂肪沉积方面,与CON+NLRP3-NC组相比,HFHC+NLRP3-NC组肝脏TG明显增高,油红O染色显示大量红色脂滴,HFHC+NLRP3-KD组肝脏TG及肝脂滴数量显著减少(P值均<0.01);在肝脏炎症方面,HFHC+NLRP3-NC组血清ALT,非酒精性脂肪性肝病活动度(NAS)评分及炎症相关基因均较CON+NLRP3-NC组明显升高,HFHC+NLRP3-KD组均明显降低(P值均<0.01);在肝纤维化方面,HFHC+NLRP3-NC组肝胶原纤维面积以及纤维化相关基因均较CON+NLRP3-NC组明显升高,HFHC+NLRP3-KD组纤维化相关基因均明显降低(P值均<0.05),胶原纤维面积虽有降低趋势但差异无统计学意义(P>0.05)。 结论 NLRP3基因敲减可显著改善高脂高糖饮食诱导的NASH小鼠模型肝脂肪沉积及炎症。 -
关键词:
- 非酒精性脂肪性肝病 /
- NLR家族, 热蛋白结构域包含蛋白3 /
- 膳食, 高脂 /
- 炎症 /
- 小鼠, 近交C57BL
Abstract:Objective To investigate the effect of NOD-like receptor family pyrin domain containing 3 (NLRP3) knockdown on a mouse model of nonalcoholic steatohepatitis (NASH) induced by high-fat high-carbohydrate (HFHC) diet. Methods A total of 44 mice were randomly divided into normal diet group (CON group) with 20 mice and HFHC group with 24 mice. At the end of week 14 of modeling, 4 mice were randomly selected from the HFHC group for the pre-experiment of adeno-associated virus (AAV) by tail vein injection, and NLRP3 knockdown was verified after 4 weeks. After NLRP3 knockdown was verified at the end of week 18, the remaining 40 mice were given a single tail vein injection of AAV, and then they were divided into CON+NLRP3 knockdown negative control group (CON+NLRP3-NC group), CON+NLRP3 knockdown group (CON+NLRP3-KD group), HFHC+NLRP3-NC group, and HFHC+NLRP3-KD group, with 10 mice in each group. At the end of week 24, the activation of NLRP3 inflammasome was observed; related indicators were measured, including body weight, liver weight, liver index, and glucose metabolism (fasting blood glucose, fasting insulin, and Homeostasis Model Assessment of Insulin Resistance [HOMA-IR] index); the indicators of liver lipid content (liver triglyceride [TG] and oil red O staining), liver inflammation (serum alanine aminotransferase [ALT] activity, HE staining, and inflammation-related genes), and liver fibrosis (Sirius Red staining and fibrosis-related genes) were measured. A one-way analysis of variance was used for comparison of continuous data between multiple groups, and the least significant difference t-test was used for further comparison between two groups. Results Compared with the CON+NLRP3-NC group based on the results of Western Blot, the HFHC+NLRP3-NC group had significant increases in the protein expression levels of NLRP3, pro-Caspase1, Caspase1, ASC, and IL-1β, while the HFHC+NLRP3-KD group had significant reductions in these levels (all P<0.05). The HFHC+NLRP3-NC group showed varying degrees of increase in body weight, liver weight, liver index, and glucose metabolism indicators, while the HFHC+NLRP3-KD group showed significant improvements in these indicators (all P<0.05). As for hepatic fat deposition, compared with the CON+NLRP3-NC group, the HFHC+NLRP3-NC group had a significant increase in liver TG, with a large number of red lipid droplets shown by oil red O staining, and the HFHC+NLRP3-KD group had significant reductions in liver TG and the number of lipid droplets in the liver (all P<0.01). In terms of liver inflammation, compared with the CON+NLRP3-NC group, the HFHC+NLRP3-NC group had significant increases in serum ALT, NAFLD activity score, and inflammation-related genes, while the HFHC+NLRP3-KD group had significant reductions in these indicators (all P<0.01). As for liver fibrosis, compared with the CON+NLRP3-NC group, the HFHC+NLRP3-NC group had significant increases in collagen fiber area and fibrosis-related genes, and the HFHC+NLRP3-KD group had significant reductions in fibrosis-related genes (all P<0.05) and a tendency of reduction in collagen fiber area (P>0.05). Conclusion NLRP3 knockdown can significantly improve hepatic fat deposition and inflammation in a mouse model of HFHC-induced NASH. -
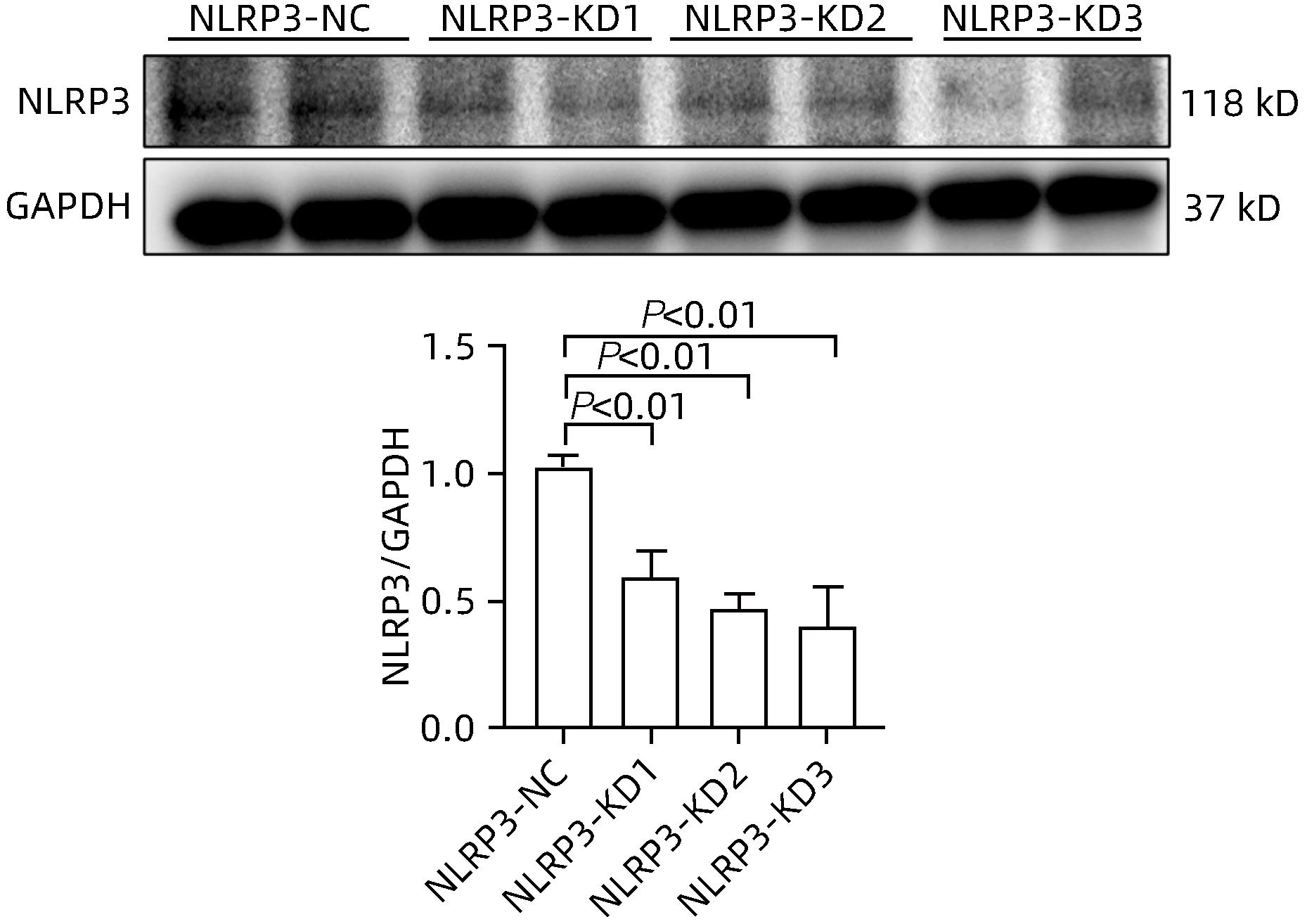
图 2 NLRP3基因敲减对肝脏NLRP3蛋白表达的影响
Figure 2. Effect of NLRP3 knockdown on liver NLRP3 protein expression

图 3 NLRP3基因敲减对肝脏NLRP3转录和蛋白水平的影响
Figure 3. Effects of NLRP3 knockdown on liver NLRP3 transcription and protein levels

图 4 NLRP3基因敲减对炎症小体相关指标及下游IL-1β蛋白的影响
Figure 4. Effects of NLRP3 knockdown on inflammatory-related proteins and IL-1β protein

图 6 NLRP3基因敲减对小鼠肝脏形态及肝脏脂滴的影响(×200)
Figure 6. Effects of NLRP3 knockdown on hepatic morphology and lipid droplets(×200)

图 7 NLRP3基因敲减对小鼠肝组织病理的影响(×200)
Figure 7. Effects of NLRP3 knockdown on hepatic histopathology (×200)

图 8 NLRP3基因敲减对小鼠肝组织胶原沉积的影响(×200)
Figure 8. Effects of NLRP3 knockdown on hepatic collagen deposition (×200)
表 1 PCR引物序列
Table 1. PCR Primer sequences
基因 上游(5'-3') 下游(5'-3') NLRP3 AACGACCCCTTCATTGAC GAGGAAGAGGAGGAAGGACA CXCL10 TCTCCATCACTCCCCTTTAC TTCGGCAGTTACTTTTGTCTC IL-1β TACATCAGCACCTCACAAGC AGAAACAGTCCAGCCCATACT CCL2 ATCGGAACCAAATGAGATCAG GCTTCAGATTTACGGGTCAAC COL1α1 TGACTGGAAGAGCGGAGAGTA GACGGCTGAGTAGGGAACAC COL3α1 CCTGGAGCCCCTGGACTAATA TCACCTGAAGGACCTCGTGTT α-SMA CTCTGCCTCTAGCACACAACT CCACGAGTAACAAATCAAAGC β-actin CCTCTATGCCAACACAGT AGCCACCAATCCACACAG 注:CXCL10,CXC趋化因子配体10;CCL2,趋化因子2;α-SMA,α平滑肌肌动蛋白;COL3α1,Ⅲ型胶原α1;COL1α1,Ⅰ型胶原α1;β-actin,β-肌动蛋白。  下载: 导出CSV
下载: 导出CSV
表 2 NLRP3基因敲减对小鼠末次肝质量、体质量、肝体比、FBG、FIN及HOMA-IR的影响
Table 2. Effects of NLRP3 knockdown on body weight, liver weight, liver/body weight ratio, FBG, FIN, and HOMA-IR
组别 动物数(只) 体质量(g) 肝质量(g) 肝体比 FBG(mg/dL) FIN(ng/mL) HOMA-IR CON+NLRP3-NC 10 26.29±1.50 0.96±0.06 0.036±0.002 4.61±0.80 0.31±0.14 0.06±0.02 CON+NLRP3-KD 10 27.27±1.42 0.98±0.07 0.038±0.004 4.13±0.64 0.41±0.16 0.07±0.02 HFHC+NLRP3-NC 10 47.09±4.401) 2.68±0.561) 0.055±0.0101) 11.41±2.361) 2.86±0.371) 1.57±0.301) HFHC+NLRP3-KD 10 43.40±5.722) 1.82±0.572) 0.041±0.0092) 10.30±2.37 2.09±0.542) 0.98±0.413) F值 70.03 32.89 11.46 41.91 80.50 52.33 P值 <0.001 <0.001 <0.001 <0.001 <0.001 <0.001 注:与CON+NLRP3-NC组相比,1) P<0.01;与HFHC+NLRP3-NC组相比,2) P<0.01,3) P<0.05。
下载: 导出CSV
表 3 NLRP3基因敲减对小鼠肝脏TG含量和油红O染色面积百分比的影响
Table 3. Effects of NLRP3 knockdown on hepatic TG content and Oil red O staining area
组别 动物数(只) 肝脏TG(mg/g) 油红O染色面积(%) CON+NLRP3-NC 10 45.65±12.97 3.82±0.80 CON+NLRP3-KD 10 54.47±20.14 1.51±0.82 HFHC+NLRP3-NC 10 119.80±21.651) 33.36±2.961) HFHC+NLRP3-KD 10 81.45±16.532) 20.72±0.902) F值 25.82 248.6 P值 <0.001 <0.001 注:与CON+NLRP3-NC组相比,1) P<0.01;与HFHC+NLRP3-NC组相比,2) P<0.01。
下载: 导出CSV
表 4 NLRP3基因敲减对小鼠血清ALT水平和肝组织NAS评分的影响
Table 4. Effects of NLRP3 knockdown on serum ALT and hepatic NAS score
组别 动物数(只) ALT(U/L) 肝细胞脂肪变性(分) 小叶内炎症(分) 肝细胞气球样变(分) NAS总分(分) CON+NLRP3-NC 10 22.03±5.56 0.00±0.00 0.00±0.00 0.00±0.00 0.00±0.00 CON+NLRP3-KD 10 14.81±1.17 0.00±0.00 0.00±0.00 0.00±0.00 0.00±0.00 HFHC+NLRP3-NC 10 174.20±71.661) 3.00±0.001) 2.67±0.521) 2.00±0.001) 7.67±0.521) HFHC+NLRP3-KD 10 67.15±47.822) 1.83±0.753) 1.33±0.523) 1.00±0.633) 4.17±1.723) F值 17.39 92.06 73.33 55.00 101.80 P值 <0.001 <0.001 <0.001 <0.001 <0.001 注:与CON+NLRP3-NC组相比,1) P<0.01;与HFHC+NLRP3-NC组相比,2) P<0.05,3) P<0.01。
下载: 导出CSV
表 5 NLRP3基因敲减对小鼠肝脏炎症相关基因表达的影响
Table 5. Effects of NLRP3 knockdown on the expression of hepatic inflammation-related genes
组别 动物数(只) IL-1β mRNA IL-6 mRNA CXCL10 mRNA CCL2 mRNA CON+NLRP3-NC 10 1.16±0.56 0.95±0.58 1.10±0.47 1.01±0.45 CON+NLRP3-KD 10 1.29±0.45 0.57±0.39 1.01±0.35 0.81±0.35 HFHC+NLRP3-NC 10 5.61±2.561) 2.01±0.611) 2.52±2.011) 2.62±1.471) HFHC+NLRP3-KD 10 3.19±1.002) 1.24±0.512) 1.32±0.412) 1.51±0.582) F值 29.09 13.10 5.73 12.93 P值 <0.001 <0.001 0.002 <0.001 注:与CON+NLRP3-NC组相比,1) P<0.01;与HFHC+NLRP3-NC组相比, 2) P<0.01。
下载: 导出CSV
表 6 NLRP3基因敲减对小鼠纤维化相关基因表达和肝脏胶原纤维的影响
Table 6. Effects of NLRP3 knockdown on the expression of fibrosis-related genes and hepatic collagen fiber
组别 动物数(只) 胶原纤维阳性面积百分比(%) mRNA 相对表达量 α-SMA mRNA COL3α1 mRNA COL1α1mRNA CON+NLRP3-NC 10 0.13±0.07 0.61±0.16 0.95±0.23 1.00±0.06 CON+NLRP3-KD 10 0.16±0.06 0.75±0.37 0.91±0.44 1.00±0.12 HFHC+NLRP3-NC 10 0.97±0.281) 0.89±0.231) 3.63±0.982) 4.08±0.532) HFHC+NLRP3-KD 10 0.61±0.16 0.56±0.183) 2.17±1.473) 2.22±0.683) F值 5.65 4.53 23.51 9.23 P值 0.006 0.007 <0.001 <0.001 注:与CON+NLRP3-NC组相比,1) P<0.05,2) P<0.01;与HFHC+NLRP3-NC组相比, 3) P<0.01。
下载: 导出CSV
-
[1] YOUNOSSI ZM, KOENIG AB, ABDELATIF D, et al. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes[J]. Hepatology, 2016, 64( 1): 73- 84. DOI: 10.1002/hep.28431. [2] YOUNOSSI Z, ANSTEE QM, MARIETTI M, et al. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention[J]. Nat Rev Gastroenterol Hepatol, 2018, 15( 1): 11- 20. DOI: 10.1038/nrgastro.2017.109. [3] DAY CP, JAMES OF. Steatohepatitis: A tale of two“hits”?[J]. Gastroenterology, 1998, 114( 4): 842- 845. DOI: 10.1016/s0016-5085(98)70599-2. [4] PRÓCHNICKI T, MANGAN MS, LATZ E. Recent insights into the molecular mechanisms of the NLRP3 inflammasome activation[J]. F1000Res, 2016, 5: F1000 Faculty Rev-1469. DOI: 10.12688/f1000research.8614.1. [5] WU XF, ZHANG F, XIONG X, et al. Tetramethylpyrazine reduces inflammation in liver fibrosis and inhibits inflammatory cytokine expression in hepatic stellate cells by modulating NLRP3 inflammasome pathway[J]. IUBMB Life, 2015, 67( 4): 312- 321. DOI: 10.1002/iub.1348. [6] YANG G, LEE HE, LEE JY. A pharmacological inhibitor of NLRP3 inflammasome prevents non-alcoholic fatty liver disease in a mouse model induced by high fat diet[J]. Sci Rep, 2016, 6: 24399. DOI: 10.1038/srep24399. [7] WEI Q, ZHU R, ZHU JY, et al. E2-induced activation of the NLRP3 inflammasome triggers pyroptosis and inhibits autophagy in HCC cells[J]. Oncol Res, 2019, 27( 7): 827- 834. DOI: 10.3727/096504018X15462920753012. [8] WANG L, HAUENSTEIN AV. The NLRP3 inflammasome: Mechanism of action, role in disease and therapies[J]. Mol Aspects Med, 2020, 76: 100889. DOI: 10.1016/j.mam.2020.100889. [9] CALCAGNO DM, CHU A, GAUL S, et al. NOD-like receptor protein 3 activation causes spontaneous inflammation and fibrosis that mimics human NASH[J]. Hepatology, 2022, 76( 3): 727- 741. DOI: 10.1002/hep.32320. [10] MRIDHA AR, WREE A, ROBERTSON AAB, et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice[J]. J Hepatol, 2017, 66( 5): 1037- 1046. DOI: 10.1016/j.jhep.2017.01.022. [11] QU JW, YUAN ZQ, WANG GY, et al. The selective NLRP3 inflammasome inhibitor MCC950 alleviates cholestatic liver injury and fibrosis in mice[J]. Int Immunopharmacol, 2019, 70: 147- 155. DOI: 10.1016/j.intimp.2019.02.016. [12] LATZ E, XIAO TS, STUTZ A. Activation and regulation of the inflammasomes[J]. Nat Rev Immunol, 2013, 13( 6): 397- 411. DOI: 10.1038/nri3452. [13] NEGRIN KA, ROTH FLACH RJ, DISTEFANO MT, et al. IL-1 signaling in obesity-induced hepatic lipogenesis and steatosis[J]. PLoS One, 2014, 9( 9): e107265. DOI: 10.1371/journal.pone.0107265. [14] QIN WW, WENG JP. Hepatocyte NLRP3 interacts with PKCε to drive hepatic insulin resistance and steatosis[J]. Sci Bull(Beijing), 2023, 68( 13): 1413- 1429. DOI: 10.1016/j.scib.2023.06.003. [15] KOHLI R, KIRBY M, XANTHAKOS SA, et al. High-fructose, medium chain trans fat diet induces liver fibrosis and elevates plasma coenzyme Q9 in a novel murine model of obesity and nonalcoholic steatohepatitis[J]. Hepatology, 2010, 52( 3): 934- 944. DOI: 10.1002/hep.23797. [16] KLEINER DE, BRUNT EM, VAN NATTA M, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease[J]. Hepatology, 2005, 41( 6): 1313- 1321. DOI: 10.1002/hep.20701. [17] VANDANMAGSAR B, YOUM YH, RAVUSSIN A, et al. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance[J]. Nat Med, 2011, 17( 2): 179- 188. DOI: 10.1038/nm.2279. [18] HUANG SL, WU YW, ZHAO ZH, et al. A new mechanism of obeticholic acid on NASH treatment by inhibiting NLRP3 inflammasome activation in macrophage[J]. Metabolism, 2021, 120: 154797. DOI: 10.1016/j.metabol.2021.154797. [19] BOARU SG, BORKHAM-KAMPHORST E, TIHAA L, et al. Expression analysis of inflammasomes in experimental models of inflammatory and fibrotic liver disease[J]. J Inflamm(Lond), 2012, 9( 1): 49. DOI: 10.1186/1476-9255-9-49. [20] CSAK T, GANZ M, PESPISA J, et al. Fatty acid and endotoxin activate inflammasomes in mouse hepatocytes that release danger signals to stimulate immune cells[J]. Hepatology, 2011, 54( 1): 133- 144. DOI: 10.1002/hep.24341. -

 本文二维码
本文二维码
计量
- 文章访问数: 1688
- HTML全文浏览量: 406
- PDF下载量: 121
- 被引次数: 0

