
 PDF下载 ( 1558 KB)
PDF下载 ( 1558 KB)

62例不明原因肝内胆汁淤积的病因谱、临床特征及基因突变分析
DOI: 10.12449/JCH250217
Etiology spectrum, clinical features, and gene mutations of unexplained intrahepatic cholestasis: An analysis of 62 cases
-
摘要:
目的 回顾性分析62例不明原因肝内胆汁淤积患者的病史、病理结果、基因测序结果,了解其病因、临床特征,以及全外显子组测序(WES)对该类疾病的诊断价值。 方法 回顾性分析2017年1月—2023年12月因不明原因肝功能异常于南京市第二医院行WES的480例患者的临床资料,并根据患者的实验室资料筛选出不明原因肝内胆汁淤积患者62例,通过影像学资料、病理表现及基因测序明确诊断。分析不明原因肝内胆汁淤积患者的人口学特征、临床表现、病因谱和基因谱。 结果 纳入患者中男35例,女27例,中位年龄42(7~77)岁。通过WES明确诊断21例(33.87%),其中家族性肝内胆汁淤积症占比最高(52.38%,11/21);通过WES排除遗传代谢相关疾病共34例,占比最高的为药物性肝损伤和脓毒症肝损伤(55.88%,19/34),原发性胆汁性胆管炎、原发性硬化性胆管炎占20.59%(7/34),肝内胆管结石占17.65%(6/34),仍无法明确诊断占11.29%(7/62)。共发现21个既往未见报道的突变位点。 结论 遗传代谢相关疾病占不明原因肝内胆汁淤积的重要部分。WES对诊断不明原因肝内胆汁淤积有重要意义。 Abstract:Objective To investigate the etiology and clinical features of intrahepatic cholestasis and the diagnostic value of whole exome sequencing (WES) through a retrospective analysis of the medical history, pathological results, and gene sequencing data of 62 patients with unexplained intrahepatic cholestasis. Methods A retrospective analysis was performed for the clinical data of 480 patients who underwent WES due to unexplained liver function abnormalities in Nanjing Second Hospital from January 2017 to December 2023, among whom 62 patients with unexplained intrahepatic cholestasis were selected based on laboratory data, and a confirmed diagnosis was made based on imaging data, pathological findings, and gene sequencing data. The patients with unexplained intrahepatic cholestasis were analyzed in terms of demographic features, clinical manifestation, etiology spectrum, and genetic profile. Results A total of 62 patients with unexplained intrahepatic cholestasis were included, among whom there were 35 male patients and 27 female patients, with a median age of 42 (7 — 77) years. WES was used to make a definite diagnosis in 21 patients (33.87%), among whom the patients with familial intrahepatic cholestasis accounted for the highest proportion of 52.38% (11/21); genetic metabolic disorders were excluded by WES in 34 patients, with drug-induced liver injury and sepsis-associated liver injury accounting for the highest proportion of 55.88% (19/34), followed by primary biliary cholangitis and primary sclerosing cholangitis accounting for 20.59% (7/34) and intrahepatic bile duct stones accounting for 17.65% (6/34), while the patients with a lack of confirmed diagnosis accounted for 11.29% (7/62). A total of 21 novel mutation sites which were not reported in previous articles were identified in this study. Conclusion Genetic metabolic disorders constitute a significant proportion of unexplained intrahepatic cholestasis, and WES plays a crucial role in the diagnosis of unexplained intrahepatic cholestasis. -
Key words:
- Liver Diseases /
- Cholestasis, Intrahepatic /
- Diagnosis /
- Mutation
-
胆汁淤积是指肝内外各种原因造成胆汁形成、分泌和排泄障碍,胆汁流不能正常流入十二指肠而进入血液的病理状态。根据其发生部位可分为肝外胆汁淤积和肝内胆汁淤积。肝内胆汁淤积病因主要有感染性、免疫性、药物和(或)毒物损伤性、遗传代谢疾病等,病因的确认常有赖于肝脏病理和基因检测[1]。早期欧洲开展一项纳入2 520例患者的多中心研究显示,初次诊断的慢性肝病患者中35%出现肝内胆汁淤积[2]。肝内胆汁淤积在慢性肝病中发生占比高,因此认识该类疾病的临床表现、病因构成,有助于明确诊断,并施以相应的治疗。
全外显子组测序(whole-exome sequencing,WES)是针对人基因组中的蛋白质编码区域进行测序,原理是利用目标序列捕获技术将基因组的全部外显子区域DNA捕获并靶向富集后进行高通量测序以发现疾病相关遗传变异,虽然外显子区域在人类全基因组中仅占1%~1.5%[3],但是目前一致认为大部分功能变异都潜藏在外显子中[4]。本研究通过对不明原因肝内胆汁淤积患者的全外显子基因测序结果进行分析,以期明确此类不明原因肝病的病因谱、临床特征及基因谱,并以了解不明原因肝内胆汁淤积的诊断流程。
1. 资料与方法
1.1 研究对象
2017年1月—2023年12月因不明原因肝病于本院就诊并进行全外显子基因测序的患者共480例。并根据患者的实验室资料筛选出不明原因肝内胆汁淤积患者62例。
1.2 纳入和排除标准
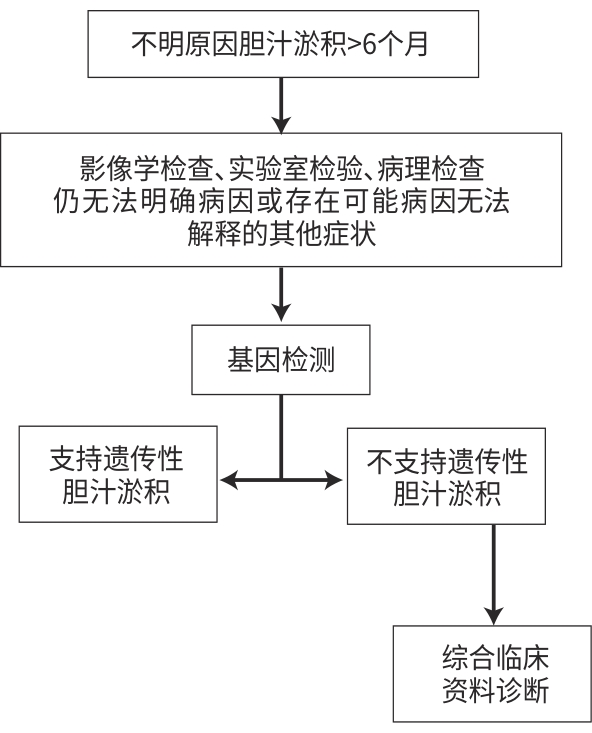
纳入标准:(1)胆汁淤积诊断标准参照欧洲肝病学会胆汁淤积性肝病治疗临床实践指南:ALP水平升高至正常值上限1.5倍以上,GGT水平升高至正常值上限3倍以上[5],和(或)总胆红素水平升高;(2)ALP水平升高至正常值上限1.5倍以上,GGT水平正常,疑似进行性家族性肝内胆汁淤积症(progressive familial intrahepatic cholestasis,PFIC)1型、2型及良性复发性肝内胆汁淤积症(benign recurrent intrahepatic cholestasis, BRIC)的患者。排除标准:(1)在胆管影像学上存在明显胆管堵塞的患者;(2)由病毒性肝炎、EB病毒、巨细胞病毒、肿瘤等引起的胆汁淤积症。不明原因肝内胆汁淤积指患者在常规无创检查(包括影像、自身抗体、免疫球蛋白、生化检查、铜蓝蛋白检查等)和肝组织病理检查后,仍不能明确病因。本研究中病因诊断严格根据疾病的指南共识及最新研究进展。若无明确的诊断标准,则根据全外显子基因测序报告,结合临床资料进行综合分析诊断。
1.3 方法
血清生物化学、病毒血清学检查;白细胞、红细胞、血红蛋白、直接抗人球蛋白试验、红细胞渗透脆性试验、血红蛋白电泳等均采用南京市第二医院检验科标准;腹部影像学检查包括电子计算机断层扫描、磁共振或彩色多普勒超声中至少1项;全外显子基因检测:基因组DNA由EDTA抗凝的外周静脉血细胞获得,使用超声波仪对DNA样品进行片段化处理,对片段化处理后的双链末端进行修复,纯化后完成DNA文库的制备,经过杂交捕获和扩增后将检测合格的文库上机测序,最后应用GATK软件、ANNOVAR软件分析变异信息并注释。肝组织活检:常规肝组织针刺活检,取长1.5~2.0 cm,直径0.1~0.2 cm肝组织,10%福尔马林固定、乙醇脱水、石蜡包埋、HE染色。病理切片由临床医生和病理医师共同读片判定。采用SPSS 25.0统计软件进行数据分析。正态分布的计量资料以
2. 结果
2.1 一般资料
本研究纳入进行全外显子基因检测的不明原因胆汁淤积性肝病患者共62例。其中,男35例 (56.45%),女27例(43.55%)。中位年龄42(7~77)岁。
2.2 临床表现
患者30.65%(19/62)表现黄疸,25.81%(16/62)表现乏力、纳差,20.97%(13/62)表现腹痛、腹胀,12.90%(8/62)有明显瘙痒表现,4.84%(3/62)表现为恶心呕吐和水肿,19.35%(12/62)仅表现为肝功能异常。
2.3 病因构成
参照全外显子基因测序结果报告,结合临床资料作出诊断,62例表现为不明原因肝内胆汁淤积患者中通过WES明确诊断的有21例(33.87%),其中家族性肝内胆汁淤积症占比最高52.38%(11/21);其余41例(66.13%)未检测到相关突变基因,回溯病史可明确诊断34例,其中占比较高的为药物/毒物性肝损伤和脓毒症肝损伤,共占55.88%(19/34),原发性胆汁性胆管炎(PBC)、原发性硬化性胆管炎(PSC)共占20.59%(7/34),肝内胆管结石占17.65%(6/34);最终仍无法明确诊断的有7例(11.29%)。62例不明原因肝内胆汁淤积病例的最终病因构成和临床特征详见表1。
表 1 62例不明原因肝内胆汁淤积病例的最终病因构成和临床特征Table 1. The final etiology and clinical characteristics of 62 cases of unexplained intrahepatic cholestasis诊断 例数 男/女(例) 年龄
(岁)
ALT
(U/L)
AST
(U/L)
ALP
(U/L)
GGT
(U/L)
TBil(μmol/L) TBA(μmol/L) 遗传代谢相关疾病 PFIC-3 8 6/2 33.00±13.05 139.89(35.10~
421.70)
150.09(34.70~
594.60)
426.83(163.40~
948.20)
500.39(226.00~
1 507.10)
46.55
(10.20~145.20)
107.96
(4.30~327.50)
BRIC-2 2 0/2 31.50±4.95 218.35
(24.70~412.00)
148.80
(40.60~257.00)
326.00
(168.00~484.00)
249.00
(15.00~483.00)
184.55
(145.30~223.80)
201.901) PFIC-1 1 1/0 27.00 9.10 22.50 167.00 31.00 215.90 256.60 ICP 1 0/1 24.00 476.00 201.70 256.00 11.90 14.20 43.80 ALGS 2 1/1 15.50±4.95 306.10
(303.40~308.80)
243.60
(239.40~247.80)
406.00
(282.00~530.00)
760.70
(152.40~
1 369.00)
25.60
(21.90~29.30)
42.75
(38.60~46.90)
Wilson病 2 1/1 13.50±2.12 149.95
(130.80~169.10)
115.05
(93.00~137.10)
335.95
(197.00~474.90)
151.00
(150.70~151.30)
22.80
(14.90~30.70)
63.00
(3.30~122.70)
ARPKD 2 1/1 45.50±7.78 33.70
(16.10~
51.30)
38.10
(17.70~58.50)
202.50
(170.00~235.00)
201.00
(176.00~226.00)
8.40
(6.70~
10.10)
13.55
(10.30~16.80)
ADPKD 1 0/1 49.00 503.60 137.60 231.00 546.00 60.20 246.80 PLD 1 0/1 42.00 56.40 46.50 211.00 312.00 13.30 11.80 17q12微缺
失综合征
1 1/0 12.00 208.90 138.20 556.00 241.00 4.20 23.20 非遗传代谢相关疾病 DILI 9 3/6 48.33±14.03 397.17
(16.60~
1 583.80)
298.69
(21.00~
1 252.90)
309.22
(174.90~529.00)
463.72
(161.00~
1 227.30)
60.34
(11.30~122.80)
66.49
(2.70~274.90)
脓毒症肝损伤 10 7/3 41.20±18.56 321.31
(18.90~
1 922.80)
183.97
(23.10~943.70)
444.46
(164.00~
1 233.00)
659.60
(128.00~
2 059.00)
86.95
(10.80~387.00)
74.64
(3.60~211.00)
PBC 4 1/3 50.50±9.26 194.95
(22.00~
469.10)
100.05
(29.90~129.30)
398.60
(188.00~656.40)
523.53
(139.00~716.00)
68.98
(7.30~210.00)
64.23
(5.30~210.00)
PSC 3 2/1 11.00(7.00~44.00) 140.17
(37.00~
246.50)
130.63
(55.20~204.10)
772.03
(241.00~
1 494.10)
324.20
(150.00~538.60)
43.17
(23.60~69.80)
52.93
(27.60~100.90)
肝内胆管结石 6 4/2 52.50(10.00~77.00) 90.35
(26.60~
150.90)
85.50
(23.70~185.20)
428.25
(224.00~654.30)
307.72
(123.00~684.00)
286.52
(10.70~908.80)
113.00
(10.10~226.80)
Budd-Chiari综合征 1 1/0 50.00 64.30 86.50 481.00 803.90 17.90 9.70 Abernethy畸形 1 0/1 51.00 810.00 448.00 221.00 199.00 148.00 未明确病因 7 6/1 46.86±17.36 54.95
(24.40~
128.60)
39.47
(20.00~
81.10)
261.21
(164.50~433.00)
318.33
(176.60~573.50)
23.10
(5.70~
38.60)
29.55
(8.40~94.20)
注:ICP,妊娠期肝内胆汁淤积症;ALGS,Alagille综合征;ARPKD,常染色体隐性多囊肾病;ADPKD,常染色体显性多囊肾病;PLD,多囊肝病;DILI,药物性肝损伤。1) 只有1例患者。
2.4 不明原因肝内胆汁淤积患者的基因检测结果
在21例通过WES检测明确诊断的患者中,共发现21个新发突变位点,基因突变信息见表2,其中有1例为片段缺失而非点突变(wes[hg19]chr17q12(34587248-36104885)X1 1.5Mb 缺失致病),未列入表中。
表 2 20例通过WES明确诊断的患者的基因突变信息Table 2. Genetic mutation information of 20 patients with a definitive diagnosis through WES病例序号 基因 核苷酸变化 氨基酸变化 ACMG评级 父亲 母亲 是否新发突变 1 JAG1 c.2990C>A Ser997* 致病性 未检查 未检查 是 2 PKHD1 c.2507T>C p.Val836Arg 可疑致病性 未检测到 杂合 是 PKHD1 c.8417T>G p.Ile2806Ser 临床意义未明 杂合 未检测到 否 3 ABCB4 c.434A>G p.Gln145Arg 临床意义未明 未检查 未检查 否 4 PKD1 c.12835C>T p.Arg4279Trp 临床意义未明 未检查 未检查 否 5 ABCB4 c.833+2T>C p.? 致病性 未检查 未检查 否 6 PKHD1 c.2312_2313insA p.Gly772fs 致病性 未检查 未检查 否 PKHD1 c.8624T>C p.Ile2875Thr 临床意义未明 未检查 未检查 否 7 ATP7B c.3809A>G p.Asn1270Ser 致病性 未检查 未检查 是 ATP7B c.2621C>T p.Ala874Val 致病性 未检查 未检查 是 8 ABCB4 c.2362C>T p.Arg788Trp 可疑致病性 未检查 未检查 是 ABCB4 c.2777C>T p.Pro926Leu 临床意义未明 未检查 未检查 否 9 ABCB4 c.3378dupT p.Ala1127fs 病理性 未检查 未检查 否 10 JAG1 c.254_258dup p.Thr87fs 病理性 未检查 未检查 否 11 ATP8B1 c.2822G>A p.Arg941Gln 临床意义未明 未检查 未检查 否 12 ALG8 c.95+5G>A p.? 临床意义未明 未检查 未检查 否 13 ABCB4 c.3250C>T p.Arg1084Trp 临床意义未明 未检查 未检查 否 14 ABCB4 c.122G>C p.Gly41Ala 临床意义未明 未检查 未检查 否 15 ATP7B c.1708-1G>C p.? 致病性 未检测到 杂合 否 ATP7B c.3316G>A p.Val1106Ile 临床意义未明 未检测到 未检测到 是 16 ABCB4 c.2362C>T p.Arg788Trp 临床意义未明 未检查 未检查 是 ABCB4 c.537-32G>T p.? 临床意义未明 未检查 未检查 否 17 ABCB11 c.239T>C p.Leu80Pro 临床意义未明 未检查 未检查 否 ATP7B c.3889G>A p.Val1297Ile 临床意义未明 未检查 未检查 否 ATP7B c.3671G>A p.Arg1224Gln 临床意义未明 未检查 未检查 否 18 ABCB11 c.1331T>C p.Val444Ala 临床意义未明 未检查 未检查 是 19 ATP8B1 c.2081T>A p.Ile694Asn 可疑致病性 未检查 未检查 是 ATP8B1 c.1288_1290del p.Lys430del 临床意义未明 未检查 未检查 否 20 ABCB4 c.1865G>A p.Gly622Glu 临床意义未明 未检查 未检查 否 2.5 不明原因肝内胆汁淤积患者的病理表现
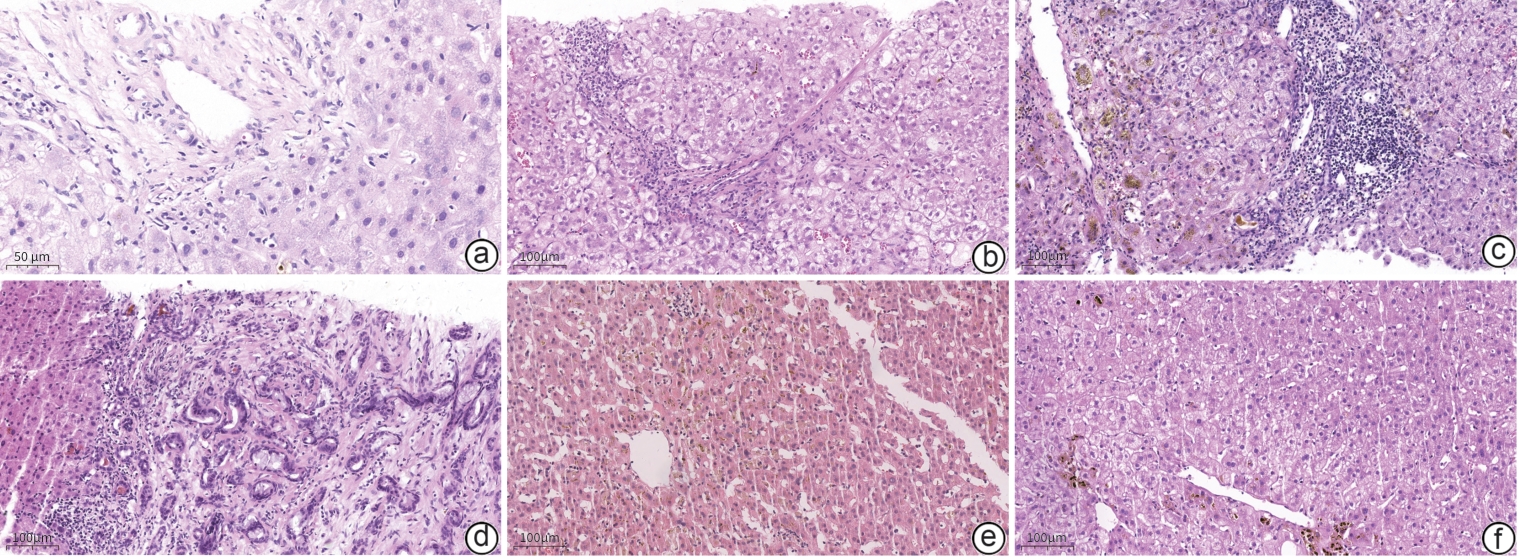
肝内胆汁淤积中常见的病理表现有:毛细胆管淤胆(常见于DILI、BRIC、ICP)、细胆管淤胆(常见于脓毒症肝损伤)、胆汁淤积性肝炎(常见于DILI)、胆管损伤(常见于PBC、PSC)、胆管板畸形[常见于先天性肝纤维化(congenital hepatic fibrosis,CHF)]。肝脏组织学表现见图1、表3。
 注: a,胆管消失综合征,为PFIC-3(×400);b,胆管损伤,为ALGS(×200);c,细胆管淤胆,为脓毒症肝损伤(×200);d,胆管板畸形,为PKD合并了CHF(×200);e,毛细胆管淤胆,为BRIC(×200);f,毛细胆管淤胆,为DILI(×200)。图 1 肝内胆汁淤积患者常见肝脏组织学表现(HE染色)Figure 1. Common histological findings in liver biopsy of patients with intrahepatic cholestasis (HE staining)表 3 常见肝内胆汁淤积的肝脏病理表现Table 3. Common hepatic pathological manifestations of intrahepatic cholestasis
注: a,胆管消失综合征,为PFIC-3(×400);b,胆管损伤,为ALGS(×200);c,细胆管淤胆,为脓毒症肝损伤(×200);d,胆管板畸形,为PKD合并了CHF(×200);e,毛细胆管淤胆,为BRIC(×200);f,毛细胆管淤胆,为DILI(×200)。图 1 肝内胆汁淤积患者常见肝脏组织学表现(HE染色)Figure 1. Common histological findings in liver biopsy of patients with intrahepatic cholestasis (HE staining)表 3 常见肝内胆汁淤积的肝脏病理表现Table 3. Common hepatic pathological manifestations of intrahepatic cholestasis疾病 病理表现 ALGS 汇管区缺乏肝小叶间胆管(胆管缺失),也无汇管区炎症和小胆管反应,肝细胞间毛细胆管缺乏CD10的表达 CHF 与PKD有关。汇管区-肝实质界面处可见许多不规则扩张的胆管(胆管板畸形)。胆管内可含浓缩的胆汁或嗜伊红物质 Wilson病 早期常见全肝小叶轻度大泡性脂肪变性和汇管区周围的肝细胞糖原核。晚期汇管区炎症加重,轻度界面炎,汇管区纤维化。与慢性胆汁淤积性肝病汇管区周围肝细胞改变相似 PFIC 表现胆汁淤积、胆管损伤、胆汁性纤维化,不同亚型PFIC病理特征不一 BRIC 表现胆汁淤积,通常不伴胆汁性纤维化,不同亚型BRIC病理表现不一 ICP 认为其与ABCB4和ABCB11等编码的蛋白缺陷有关。毛细胆管扩张,肝细胞和Kupffer细胞内可见胆红素淤积 脓毒症 浓缩的胆汁淤积导致胆管扩张(胆管性淤积),扩张的毛细胆管内可见大量的胆栓栓子 药物诱导的肝内
胆汁淤积
合成类固醇诱导的单纯性淤胆:肝细胞和毛细胆管显著淤胆,不伴有明显活动性炎症;药物诱导的急性淤胆型肝炎:中央静脉周围坏死伴有明显的肝细胞和毛细胆管胆汁淤积 PBC 可存在不同程度的胆管损伤,损伤胆管被密集的淋巴组织细胞浸润的旺炽性胆管病变是PBC突出病理特点。早期PBC的毛细胆管淤胆不常见,直到PBC晚期才会形成 PSC 胆管树的炎症、狭窄及囊性扩张为特点。胆管周围水肿、同心圆纤维化、细胆管增生、汇管区炎症、小胆管萎缩或消失也提示该病 注:PKD,多囊肾病。
3. 讨论
肝内胆汁淤积的临床表现轻重不一,病因各异,明确诊断仍是临床较为棘手的问题。本研究旨在分析不明原因肝内胆汁淤积的病因、临床特征,以及了解WES在明确病因诊断中的作用,以期提高临床医师对该类疾病的认识。
本研究通过对62例不明原因肝内胆汁淤积行WES的患者进行分析,通过WES明确诊断为遗传代谢性相关肝病21例,其中占比最高的为家族性肝内胆汁淤积症,其次为PKD、ALGS、Wilson病、ICP、PLD、17q12微缺失综合征。
PFIC是一种常染色体隐性遗传的胆汁淤积性肝病[6-7],而PFIC-3与PFIC-1/2不同,PFIC-3好发于青少年或成年人[8],因PFIC-3缺乏特异的临床表现与体征,明确诊断依赖于ABCB4基因检测。本研究9例PFIC患者年龄最大54岁,最小18岁,临床表现为慢性胆汁淤积,肝脏病理均有汇管区胆管减少或缺失的表现,ABCB4、ABCB11基因检测均检出致病性突变,其中c.2362C>T (p.Arg788Trp)已有报道[9],其余变异未见报道,其中1例PFIC-2的c.2081T>A(p.Ile694Asn)已见发表。BRIC通常被认为属于家族性胆汁淤积症的一部分,与PFIC相同,BRIC也有不同的亚型,与PFIC相应亚型的致病基因相同。但是BRIC的临床表现与PFIC不尽相同,BRIC常呈特发性、持续数周到数年不等的胆汁淤积,而后完全消退,预后良好,通常不会导致肝纤维化[10]。本研究中2例BRIC患者临床表现为较严重的胆汁淤积症,均存在明显的黄疸、严重的皮肤瘙痒;但病理表现炎症轻微,无纤维化,可见明显的胆栓形成及3区为主的淤胆,ABCB11突变均为新发现突变。ICP是一种发生于妊娠中晚期的重要产科并发症。ICP最初的主要症状为发生在妊娠中晚期(通常在妊娠30周以后)的皮肤瘙痒[11]。国际上对于ICP的实验室诊断至今尚无统一标准,ICP临床诊治和管理指南推荐空腹血清TBA≥10 μmol/L或餐后血清TBA≥19 μmol/L作为ICP的诊断标准[12]。有研究指出ICP可能与ATP8B1基因、ABCB11基因、ABCB4基因和由NR1H4基因编码的FXR基因变异有关[13]。本研究中的ICP患者为孕周为28周的产妇,临床表现为剧烈皮肤瘙痒,行WES发现ABCB11与ABCB4基因突变ABCB11 c.3084A>G(p.Ala1028=)、ABCB11 c.1331T>C(p.Val444Ala)、ABCB4 c.711A>T(p.Ile237=)、ABCB4 c.175C>T(p.Leu59=),有文献指出,ABCB11中常见的p.Val444Ala突变体可增加DILI和ICP的风险[14],因此诊断ICP。ALGS为儿科最常见的遗传性胆汁淤积症,是涉及多器官的常染色体显性遗传病,发病率约为1/30 000[15]。ALGS临床表现较为特异,累及多系统。实验室检查常表现为ALP、GGT升高,GGT升高更为明显。75%~90%患者肝脏病理组织学表现为胆管缺失。其致病基因大多数为JAG1基因突变,少数为NOTCH2基因突变[15],本研究2例ALGS患者,分别为12岁、19岁,临床表现符合诊断,肝穿病理均表现为多个汇管区胆管缺失。WES结果显示两者均为JAG1基因突变,可明确诊断。其中c.254_258dup(p.Thr87fs)为新发现的突变位点,另一突变位点在中国ALGS患者中已有报道[16]。Wilson病是一种由ATP7B基因突变引起的铜代谢常染色体隐性遗传病。全球范围内ATP7B基因突变携带率为1/90,Wilson病患病率约0.25/10 000~4/10 000[17]。Wilson病的肝病表现缺乏特异性,如角膜K-F环、血清铜降低与肝铜沉积也可继发于其他慢性胆汁淤积性肝病[18],因此临床表现为胆汁淤积的Wilson病诊断的明确仍需要ATP7B基因检测。本研究中2例Wilson病患者根据指南明确诊断[17],分别为12岁与15岁,其中1例临床表现较重,出现明显黄疸、双下肢水肿等肝硬化表现,另一例则无明显临床症状,仅表现为GGT与ALP水平升高,ATP7B基因检测所检出的c.3316G>A(p.Val1106lle)、c.3809A>G(p.Asn1270Ser)、c.2621C>T(p.Ala874Val)已有报道,余为新发现突变。PKD常与CHF共存,肝脏病理活检存在胆管板畸形的重要病理特征,CHF发病机制仍不清楚[19]。本研究中PKD患者临床表现为腹痛、腹胀等消化道症状,肾脏超声影像学表现符合诊断标准[20],肝穿病理提示胆管板畸形,进一步行WES检出PKHD1和PKD1基因突变,明确诊断。PLD是一种常染色体显性遗传疾病,主要特点是胆管上皮细胞的过度生长,表现为肝实质多个弥散分布的囊性病变,常与成人多囊肾伴发[21],有文献指出ALG8基因编码的蛋白突变可通过损害多囊蛋白-1的翻译后修饰和转运至细胞膜,从而可以改变囊性疾病的严重程度[22]。本研究中该患者为中年女性,反复肝功能异常7年余,否认用药史、饮酒史等,肝炎全套检测阴性,超声影像仅发现数个肝脏囊肿,行肝穿为基本正常肝组织,遂行WES发现ALG8基因突变,RDDC数据库预测为致病突变,考虑PLD。17q12微缺失综合征(17q12 Microdeletion syndrome)与肾囊肿、青少年起病的成人型糖尿病有关,有文献报道17q12微缺失综合征患儿肝脏病理活检存在胆管缺失和胆汁淤积,眼科检查存在角膜后胚胎环,呈现ALGS表现[23]。本研究中患者临床表现为乏力、纳差、黄疸,胆系酶升高,WES检出17q12区域1.5Mb缺失,明确诊断为17q12微缺失综合征。
41例未检测到突变基因的患者,回溯病史可明确诊断34例,仍有7例无法明确诊断。确诊的疾病中主要为DILI、脓毒症肝损伤、自身免疫性肝病、肝内胆管结石。WES在诊断程序中起到了排除遗传相关胆汁淤积的作用。
本研究中的DILI患者的明确诊断参照《中国药物性肝损伤诊治指南(2023年版)》[24]。10例脓毒症肝损伤患者临床可明确存在感染,肝脏病理发现汇管区周围细胆管淤胆,提示脓毒症肝损伤,感染控制后持续黄疸或胆系酶高,经WES排除遗传代谢相关肝病可确诊。本研究中的4例PBC病例为不典型PBC,病理表现均存在胆管减少或胆管损伤,但其中1例肝脏病理学表现存在不符合病程的毛细胆管淤胆,2例存在间接胆红素升高,1例影像学表现存在多个肝囊肿,遂行WES明确是否存在其他遗传代谢相关肝病。本研究中3例PSC患者临床表现为胆汁淤积,均存在腹痛、腹胀、乏力等症状,但影像学未发现胆管系统多灶性狭窄、节段性扩张、串珠样改变及枯树枝样改变,肝脏病理表现小叶间胆管基底膜增厚,但未见“洋葱皮样”胆管周围纤维化改变,遂行WES排除遗传代谢相关疾病,其在后续随访中明确诊断为PSC。肝胆管结石病特指始发于肝内胆管系统的结石。因单一的检查常不能获得全面的诊断,往往需要一种以上的影像学检查相互印证才能达到正确诊断的目的[25]。遂在行侵入性检查之前先行WES排除是否存在遗传代谢相关肝病,后续行侵入性检查明确为肝内胆管结石。本研究中也发现2例可能由血管相关肝病引起的肝内胆汁淤积,本研究中Abernethy畸形及Budd-Chiari综合征由影像学确诊,既往有个案报道其临床表现可表现为胆汁淤积[26-27]。
本研究中仍有7例无法明确诊断,可能有以下原因:(1)临床病史不完善,既往史未明确;(2)突变发生在WES未检测到的区域(一项对882个家庭队列进行基因测序的研究显示,全外显子仍有8%的漏诊率[28]);(3)大部分患者未行家系分析,家系分析有助于帮助患者确定是否遗传家族某些疾病的风险。
综上,不明原因胆汁淤积病因复杂,遗传代谢相关肝病是不明原因胆汁淤积性肝病的重要组成部分,诊断思路可参考图2。不明原因肝内胆汁淤积性临床表现类似,病理表现不尽相似,明确诊断仍是一大难题,WES对遗传代谢相关肝病的诊断非常关键。
-

注: a,胆管消失综合征,为PFIC-3(×400);b,胆管损伤,为ALGS(×200);c,细胆管淤胆,为脓毒症肝损伤(×200);d,胆管板畸形,为PKD合并了CHF(×200);e,毛细胆管淤胆,为BRIC(×200);f,毛细胆管淤胆,为DILI(×200)。
图 1 肝内胆汁淤积患者常见肝脏组织学表现(HE染色)
Figure 1. Common histological findings in liver biopsy of patients with intrahepatic cholestasis (HE staining)
表 1 62例不明原因肝内胆汁淤积病例的最终病因构成和临床特征
Table 1. The final etiology and clinical characteristics of 62 cases of unexplained intrahepatic cholestasis
诊断 例数 男/女(例) 年龄
(岁)
ALT
(U/L)
AST
(U/L)
ALP
(U/L)
GGT
(U/L)
TBil(μmol/L) TBA(μmol/L) 遗传代谢相关疾病 PFIC-3 8 6/2 33.00±13.05 139.89(35.10~
421.70)
150.09(34.70~
594.60)
426.83(163.40~
948.20)
500.39(226.00~
1 507.10)
46.55
(10.20~145.20)
107.96
(4.30~327.50)
BRIC-2 2 0/2 31.50±4.95 218.35
(24.70~412.00)
148.80
(40.60~257.00)
326.00
(168.00~484.00)
249.00
(15.00~483.00)
184.55
(145.30~223.80)
201.901) PFIC-1 1 1/0 27.00 9.10 22.50 167.00 31.00 215.90 256.60 ICP 1 0/1 24.00 476.00 201.70 256.00 11.90 14.20 43.80 ALGS 2 1/1 15.50±4.95 306.10
(303.40~308.80)
243.60
(239.40~247.80)
406.00
(282.00~530.00)
760.70
(152.40~
1 369.00)
25.60
(21.90~29.30)
42.75
(38.60~46.90)
Wilson病 2 1/1 13.50±2.12 149.95
(130.80~169.10)
115.05
(93.00~137.10)
335.95
(197.00~474.90)
151.00
(150.70~151.30)
22.80
(14.90~30.70)
63.00
(3.30~122.70)
ARPKD 2 1/1 45.50±7.78 33.70
(16.10~
51.30)
38.10
(17.70~58.50)
202.50
(170.00~235.00)
201.00
(176.00~226.00)
8.40
(6.70~
10.10)
13.55
(10.30~16.80)
ADPKD 1 0/1 49.00 503.60 137.60 231.00 546.00 60.20 246.80 PLD 1 0/1 42.00 56.40 46.50 211.00 312.00 13.30 11.80 17q12微缺
失综合征
1 1/0 12.00 208.90 138.20 556.00 241.00 4.20 23.20 非遗传代谢相关疾病 DILI 9 3/6 48.33±14.03 397.17
(16.60~
1 583.80)
298.69
(21.00~
1 252.90)
309.22
(174.90~529.00)
463.72
(161.00~
1 227.30)
60.34
(11.30~122.80)
66.49
(2.70~274.90)
脓毒症肝损伤 10 7/3 41.20±18.56 321.31
(18.90~
1 922.80)
183.97
(23.10~943.70)
444.46
(164.00~
1 233.00)
659.60
(128.00~
2 059.00)
86.95
(10.80~387.00)
74.64
(3.60~211.00)
PBC 4 1/3 50.50±9.26 194.95
(22.00~
469.10)
100.05
(29.90~129.30)
398.60
(188.00~656.40)
523.53
(139.00~716.00)
68.98
(7.30~210.00)
64.23
(5.30~210.00)
PSC 3 2/1 11.00(7.00~44.00) 140.17
(37.00~
246.50)
130.63
(55.20~204.10)
772.03
(241.00~
1 494.10)
324.20
(150.00~538.60)
43.17
(23.60~69.80)
52.93
(27.60~100.90)
肝内胆管结石 6 4/2 52.50(10.00~77.00) 90.35
(26.60~
150.90)
85.50
(23.70~185.20)
428.25
(224.00~654.30)
307.72
(123.00~684.00)
286.52
(10.70~908.80)
113.00
(10.10~226.80)
Budd-Chiari综合征 1 1/0 50.00 64.30 86.50 481.00 803.90 17.90 9.70 Abernethy畸形 1 0/1 51.00 810.00 448.00 221.00 199.00 148.00 未明确病因 7 6/1 46.86±17.36 54.95
(24.40~
128.60)
39.47
(20.00~
81.10)
261.21
(164.50~433.00)
318.33
(176.60~573.50)
23.10
(5.70~
38.60)
29.55
(8.40~94.20)
注:ICP,妊娠期肝内胆汁淤积症;ALGS,Alagille综合征;ARPKD,常染色体隐性多囊肾病;ADPKD,常染色体显性多囊肾病;PLD,多囊肝病;DILI,药物性肝损伤。1) 只有1例患者。
 下载: 导出CSV
下载: 导出CSV
表 2 20例通过WES明确诊断的患者的基因突变信息
Table 2. Genetic mutation information of 20 patients with a definitive diagnosis through WES
病例序号 基因 核苷酸变化 氨基酸变化 ACMG评级 父亲 母亲 是否新发突变 1 JAG1 c.2990C>A Ser997* 致病性 未检查 未检查 是 2 PKHD1 c.2507T>C p.Val836Arg 可疑致病性 未检测到 杂合 是 PKHD1 c.8417T>G p.Ile2806Ser 临床意义未明 杂合 未检测到 否 3 ABCB4 c.434A>G p.Gln145Arg 临床意义未明 未检查 未检查 否 4 PKD1 c.12835C>T p.Arg4279Trp 临床意义未明 未检查 未检查 否 5 ABCB4 c.833+2T>C p.? 致病性 未检查 未检查 否 6 PKHD1 c.2312_2313insA p.Gly772fs 致病性 未检查 未检查 否 PKHD1 c.8624T>C p.Ile2875Thr 临床意义未明 未检查 未检查 否 7 ATP7B c.3809A>G p.Asn1270Ser 致病性 未检查 未检查 是 ATP7B c.2621C>T p.Ala874Val 致病性 未检查 未检查 是 8 ABCB4 c.2362C>T p.Arg788Trp 可疑致病性 未检查 未检查 是 ABCB4 c.2777C>T p.Pro926Leu 临床意义未明 未检查 未检查 否 9 ABCB4 c.3378dupT p.Ala1127fs 病理性 未检查 未检查 否 10 JAG1 c.254_258dup p.Thr87fs 病理性 未检查 未检查 否 11 ATP8B1 c.2822G>A p.Arg941Gln 临床意义未明 未检查 未检查 否 12 ALG8 c.95+5G>A p.? 临床意义未明 未检查 未检查 否 13 ABCB4 c.3250C>T p.Arg1084Trp 临床意义未明 未检查 未检查 否 14 ABCB4 c.122G>C p.Gly41Ala 临床意义未明 未检查 未检查 否 15 ATP7B c.1708-1G>C p.? 致病性 未检测到 杂合 否 ATP7B c.3316G>A p.Val1106Ile 临床意义未明 未检测到 未检测到 是 16 ABCB4 c.2362C>T p.Arg788Trp 临床意义未明 未检查 未检查 是 ABCB4 c.537-32G>T p.? 临床意义未明 未检查 未检查 否 17 ABCB11 c.239T>C p.Leu80Pro 临床意义未明 未检查 未检查 否 ATP7B c.3889G>A p.Val1297Ile 临床意义未明 未检查 未检查 否 ATP7B c.3671G>A p.Arg1224Gln 临床意义未明 未检查 未检查 否 18 ABCB11 c.1331T>C p.Val444Ala 临床意义未明 未检查 未检查 是 19 ATP8B1 c.2081T>A p.Ile694Asn 可疑致病性 未检查 未检查 是 ATP8B1 c.1288_1290del p.Lys430del 临床意义未明 未检查 未检查 否 20 ABCB4 c.1865G>A p.Gly622Glu 临床意义未明 未检查 未检查 否
下载: 导出CSV
表 3 常见肝内胆汁淤积的肝脏病理表现
Table 3. Common hepatic pathological manifestations of intrahepatic cholestasis
疾病 病理表现 ALGS 汇管区缺乏肝小叶间胆管(胆管缺失),也无汇管区炎症和小胆管反应,肝细胞间毛细胆管缺乏CD10的表达 CHF 与PKD有关。汇管区-肝实质界面处可见许多不规则扩张的胆管(胆管板畸形)。胆管内可含浓缩的胆汁或嗜伊红物质 Wilson病 早期常见全肝小叶轻度大泡性脂肪变性和汇管区周围的肝细胞糖原核。晚期汇管区炎症加重,轻度界面炎,汇管区纤维化。与慢性胆汁淤积性肝病汇管区周围肝细胞改变相似 PFIC 表现胆汁淤积、胆管损伤、胆汁性纤维化,不同亚型PFIC病理特征不一 BRIC 表现胆汁淤积,通常不伴胆汁性纤维化,不同亚型BRIC病理表现不一 ICP 认为其与ABCB4和ABCB11等编码的蛋白缺陷有关。毛细胆管扩张,肝细胞和Kupffer细胞内可见胆红素淤积 脓毒症 浓缩的胆汁淤积导致胆管扩张(胆管性淤积),扩张的毛细胆管内可见大量的胆栓栓子 药物诱导的肝内
胆汁淤积
合成类固醇诱导的单纯性淤胆:肝细胞和毛细胆管显著淤胆,不伴有明显活动性炎症;药物诱导的急性淤胆型肝炎:中央静脉周围坏死伴有明显的肝细胞和毛细胆管胆汁淤积 PBC 可存在不同程度的胆管损伤,损伤胆管被密集的淋巴组织细胞浸润的旺炽性胆管病变是PBC突出病理特点。早期PBC的毛细胆管淤胆不常见,直到PBC晚期才会形成 PSC 胆管树的炎症、狭窄及囊性扩张为特点。胆管周围水肿、同心圆纤维化、细胆管增生、汇管区炎症、小胆管萎缩或消失也提示该病 注:PKD,多囊肾病。
下载: 导出CSV
-
[1] Chinese Society of Hepatology, Chinese Medical Association. Guidelines on the management of cholestasis liver diseases(2021)[J]. J Clin Hepatol, 2022, 38( 1): 62- 69. DOI: 10.3969/j.issn.1001-5256.2022.01.010.中华医学会肝病学分会. 胆汁淤积性肝病管理指南(2021)[J]. 临床肝胆病杂志, 2022, 38( 1): 62- 69. DOI: 10.3969/j.issn.1001-5256.2022.01.010. [2] BORTOLINI M, ALMASIO P, BRAY G, et al. Multicentre survey of the prevalence of intrahepatic cholestasis in 2520 consecutive patients with newly diagnosed chronic liver disease[J]. Drug Investig, 1992, 4( 4): 83- 89. DOI: 10.1007/BF03258368. [3] NG SB, BUCKINGHAM KJ, LEE C, et al. Exome sequencing identifies the cause of a Mendelian disorder[J]. Nat Genet, 2010, 42( 1): 30- 35. DOI: 10.1038/ng.499. [4] BOTSTEIN D, RISCH N. Discovering genotypes underlying human phenotypes: Past successes for Mendelian disease, future approaches for complex disease[J]. Nat Genet, 2003, 33 Suppl: 228- 237. DOI: 10.1038/ng1090. [5] European Association for the Study of the Liver. EASL Clinical Practice Guidelines: Management of cholestatic liver diseases[J]. J Hepatol, 2009, 51( 2): 237- 267. DOI: 10.1016/j.jhep.2009.04.009. [6] GUNAYDIN M, BOZKURTER CIL AT. Progressive familial intrahepatic cholestasis: Diagnosis, management, and treatment[J]. Hepat Med, 2018, 10: 95- 104. DOI: 10.2147/HMER.S137209. [7] GUO J, XIE SY, YANG LR, et al. Clinical characteristics of 2 cases of progressive familial intrahepatic cholestasis and literature review[J/CD]. Chin J Liver Dis(Electronic Version), 2024, 16( 1): 67- 72. DOI: 10.3969/j.issn.1674-7380.2024.01.012.郭静, 谢双宇, 杨玲蓉, 等. 进行性家族性肝内胆汁淤积症2例临床特点分析及文献复习[J/CD]. 中国肝脏病杂志(电子版), 2024, 16( 1): 67- 72. DOI: 10.3969/j.issn.1674-7380.2024.01.012. [8] WENG YH, XIONG QF, LIU DX, et al. Clinical and pathological features of progressive familial intrahepatic cholestasis type 3[J]. J Clin Hepatol, 2022, 38( 1): 154- 159. DOI: 10.3969/j.issn.1001-5256.2022.01.024.翁宇航, 熊清芳, 刘杜先, 等. 进行性家族性肝内胆汁淤积症3型临床病理特征分析[J]. 临床肝胆病杂志, 2022, 38( 1): 154- 159. DOI: 10.3969/j.issn.1001-5256.2022.01.024. [9] GOTTHARDT D, RUNZ H, KEITEL V, et al. A mutation in the canalicular phospholipid transporter gene, ABCB4 is associated with cholestasis, ductopenia, and cirrhosis in adults[J]. Hepatology, 2008, 48( 4): 1157- 1166. DOI: 10.1002/hep.22485. [10] HALAWI A, IBRAHIM N, BITAR R. Triggers of benign recurrent intrahepatic cholestasis and its pathophysiology: A review of literature[J]. Acta Gastroenterol Belg, 2021, 84( 3): 477- 486. DOI: 10.51821/84.3.013. [11] CHEN FF, PENG LQ, WANG J. Correlation between serum glycocholic acid and liver biochemical indexes and inflammatory cytokines in patients with intrahepatic chole-stasis of pregnancy[J]. J Clin Exp Med, 2023, 22( 14): 1534- 1537. DOI: 10.3969/j.issn.1671-4695.2023.14.021.陈福芳, 彭丽钦, 汪洁. 妊娠期肝内胆汁淤积症患者血清甘胆酸与肝生化指标、炎性细胞因子水平变化相关性分析[J]. 临床和实验医学杂志, 2023, 22( 14): 1534- 1537. DOI: 10.3969/j.issn.1671-4695.2023.14.021. [12] Obstetrics Subgroup, Society of Obstetrics and Gynecology, Chinese Medical Association; Society of Perinatal Medicine, Chinese Medical Association. Guidelines for clinical diagnosis, treatment and management of intrahepatic cholestasis of pregnancy(2024)[J]. Chin J Obstet Gynecol, 2024, 59( 2): 97- 107. DOI: 10.3760/cma.j.cn112141-20230914-00099.中华医学会妇产科学分会产科学组, 中华医学会围产医学分会. 妊娠期肝内胆汁淤积症临床诊治和管理指南(2024版)[J]. 中华妇产科杂志, 2024, 59( 2): 97- 107. DOI: 10.3760/cma.j.cn112141-20230914-00099. [13] AYDıN GA, ÖZGEN G, GÖRÜKMEZ O. The role of genetic mutations in intrahepatic cholestasis of pregnancy[J]. Taiwan J Obstet Gynecol, 2020, 59( 5): 706- 710. DOI: 10.1016/j.tjog.2020.07.014. [14] NAYAGAM JS, WILLIAMSON C, JOSHI D, et al. Review article: Liver disease in adults with variants in the cholestasis-related genes ABCB11 ABCB4 and ATP8B1[J]. Aliment Pharmacol Ther, 2020, 52( 11-12): 1628- 1639. DOI: 10.1111/apt.16118. [15] AYOUB MD, KAMATH BM. Alagille syndrome: Current understanding of pathogenesis, and challenges in diagnosis and management[J]. Clin Liver Dis, 2022, 26( 3): 355- 370. DOI: 10.1016/j.cld.2022.03.002. [16] LI LT, DONG JB, WANG XH, et al. JAG1 mutation spectrum and origin in Chinese children with clinical features of alagille syndrome[J]. PLoS One, 2015, 10( 6): e0130355. DOI: 10.1371/journal.pone.0130355. [17] Inherited Metabolic Liver Disease Collaboration Group, Chinese Society of Hepatology, Chinese Medical Association. Guidelines for the diagnosis and treatment of hepatolenticular degeneration(2022 edition)[J]. Chin J Hepatol, 2022, 30( 1): 9- 20. DOI: 10.3760/cma.j.cn501113-20211217-00603.中华医学会肝病学分会遗传代谢性肝病协作组. 肝豆状核变性诊疗指南(2022年版)[J]. 中华肝脏病杂志, 2022, 30( 1): 9- 20. DOI: 10.3760/cma.j.cn501113-20211217-00603. [18] DANKS DM. Copper and liver disease[J]. Eur J Pediatr, 1991, 150( 3): 142- 148. DOI: 10.1007/BF01963553. [19] HASBAOUI BE, RIFAI Z, SAGHIR S, et al. Congenital hepatic fibrosis: Case report and review of literature[J]. Pan Afr Med J, 2021, 38: 188. DOI: 10.11604/pamj.2021.38.188.27941. [20] Writing Group For Practice Guidelines For Diagnosis And Treatment Of Genetic Diseases Medical Genetics Branch Of Chinese Medical Association. Clinical practice guidelines for polycystic kidney diseases[J]. Chin J Med Genet, 2020, 37( 3): 277- 283. DOI: 10.3760/cma.j.issn.1003-9406.2020.03.009.中华医学会医学遗传学分会遗传病临床实践指南撰写组. 多囊肾病的临床实践指南[J]. 中华医学遗传学杂志, 2020, 37( 3): 277- 283. DOI: 10.3760/cma.j.issn.1003-9406.2020.03.009. [21] KARHUNEN PJ, TENHU M. Adult polycystic liver and kidney diseases are separate entities[J]. Clin Genet, 1986, 30( 1): 29- 37. DOI: 10.1111/j.1399-0004.1986.tb00565.x. [22] LANKTREE MB, HAGHIGHI A, di BARI I, et al. Insights into autosomal dominant polycystic kidney disease from genetic studies[J]. Clin J Am Soc Nephrol, 2021, 16( 5): 790- 799. DOI: 10.2215/CJN.02320220. [23] DIXIT A, PATEL C, HARRISON R, et al. 17q12 microdeletion syndrome: Three patients illustrating the phenotypic spectrum[J]. Am J Med Genet A, 2012, 158 A( 9): 2317- 2321. DOI: 10.1002/ajmg.a.35520. [24] Technology Committee on DILI Prevention and Management, Chinese Medical Biotechnology Association; Study Group of Drug-Induced Liver Disease, Chinese Medical Association for the Study of Liver Diseases. Chinese guideline for diagnosis and management of drug-induced liver injury(2023 version)[J]. Chin J Gastroenterol, 2023, 28( 7): 397- 431. DOI: 10.3760/cma.j.cn501113-20230419-00176.中国医药生物技术协会药物性肝损伤防治技术专业委员会, 中华医学会肝病学分会药物性肝病学组. 中国药物性肝损伤诊治指南(2023年版)[J]. 胃肠病学, 2023, 28( 7): 397- 431. DOI: 10.3760/cma.j.cn501113-20230419-00176. [25] Biliary Surgery Group of the Surgery Branch of the Chinese Medical Association. Guidelines for diagnosis and treatment of hepatolithiasis[J]. Chin J Dig Surg, 2007, 6( 2): 156- 161. DOI: 10.3760/cma.j.issn.1673-9752.2007.02.028.中华医学会外科学分会胆道外科学组. 肝胆管结石病诊断治疗指南[J]. 中华消化外科杂志, 2007, 6( 2): 156- 161. DOI: 10.3760/cma.j.issn.1673-9752.2007.02.028. [26] YAO X, LIU Y, YU LD, et al. Rare portal hypertension caused by abernethy malformation(type IIC): A case report[J]. World J Radiol, 2023, 15( 8): 250- 255. DOI: 10.4329/wjr.v15.i8.250. [27] ENNAIFER R, BACHA D, ROMDHANE H, et al. Budd-chiari syndrome: An unusual presentation of multisystemic sarcoidosis[J]. Clin Pract, 2015, 5( 3): 768. DOI: 10.4081/cp.2015.768. [28] WOJCIK MH, LEMIRE G, BERGER E, et al. Genome sequencing for diagnosing rare diseases[J]. N Engl J Med, 2024, 390( 21): 1985- 1997. DOI: 10.1056/NEJMoa2314761. -

 下载:
下载:

 本文二维码
本文二维码
计量
- 文章访问数: 2090
- HTML全文浏览量: 89
- PDF下载量: 33
- 被引次数: 0

