
 PDF下载 ( 1904 KB)
PDF下载 ( 1904 KB)

原发性胆汁性胆管炎合并结缔组织病及免疫性血小板减少症1例报告
DOI: 10.3969/j.issn.1001-5256.2023.06.022
A case of primary biliary cholangitis with connective tissue disease and immune thrombocytopenia
-
-
Key words:
- Primary Biliary Cholangitis /
- Connective Tissue Disease /
- Thrombocytopenia
-
原发性胆汁性胆管炎(PBC)可合并多种肝外自身免疫性疾病,包括甲状腺炎、溃疡性结肠炎、干燥综合征、类风湿性关节炎、系统性红斑狼疮、免疫性血小板减少症(Idiopathic thrombocytopenic purpura,ITP)等。在此报道1例PBC合并结缔组织病及ITP的病例,结合文献讨论,提高临床医生对该病的认识。
1. 病例资料
患者女性,36岁,因“双上肢散在瘀斑1周,鼻衄1天”于2022年9月20日入本院诊治。患者2022年9月13日无明显诱因出现双上肢散在瘀斑,未在意,未系统诊治。2022年9月19日就诊于白山市妇幼保健院,行血常规检查:血红蛋白78 g/L,血小板1.0×109/L,就诊途中出现鼻衄,出血量较少,无法自行止血,于当地医院急诊,行填塞止血治疗,后未再出血。2022年9月20日为求进一步系统诊治遂来本院。病程中患者自述无发热、盗汗,无头晕、头痛,无咳嗽、咳痰、胸痛、呼吸困难,无腹痛、腹泻,无尿频、尿急、尿痛,无指(趾)端苍白、发绀、潮红,无关节痛,无皮疹,近期体质量未见明显下降。追问病史,自述近期月经量增多,经期延长,既往体健,妊娠4次,顺产2胎,流产2胎。查体:身高165 cm,体质量70 kg,生命体征平稳,神志清楚,贫血貌,巩膜无黄染,上肢输液部位可见片状瘀斑,双上肢、下肢可见散在瘀斑,两侧对称性分布,与周围分界清楚,不高于皮面,压之不褪色,无红肿、疼痛。未触及肿大淋巴结,心肺查体无异常,腹壁可见散在出血点,腹软,无压痛、反跳痛及肌紧张,肝脾肋下未触及,双下肢无水肿。
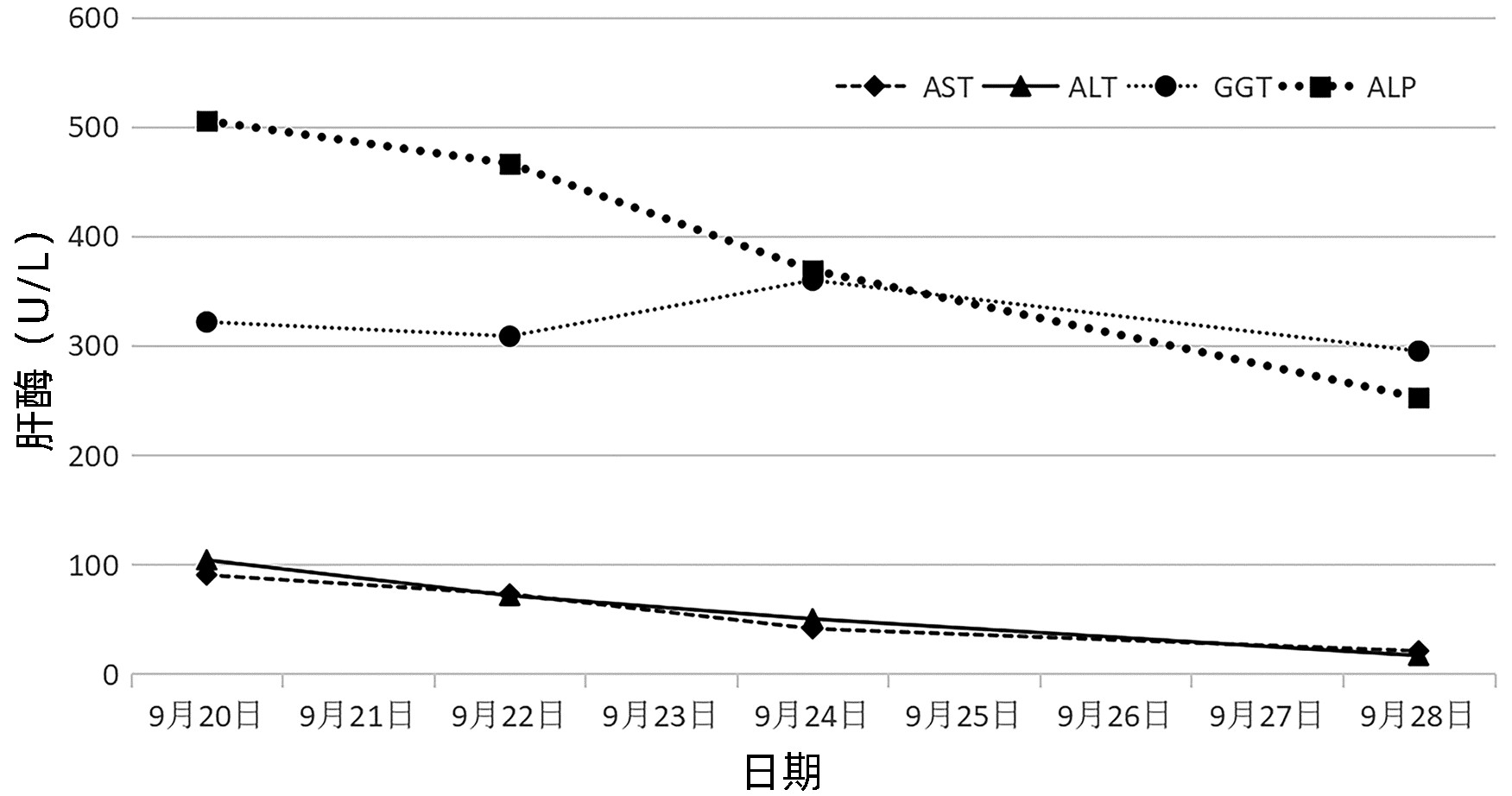
行辅助检查,血常规:血小板5.0×109/L,血红蛋白74 g/L,网织红细胞百分比2.43%,网织红细胞绝对值0.092 1×1012/L,未成熟网织红细胞比率22.60%,网织红细胞血红蛋白含量17.5 pg,低荧光网织红细胞比率77.40%,高荧光网织红细胞比率8.50%;血沉120 mm/h。甲状腺功能检查:甲状腺球蛋白抗体(A-Tg)291.77 IU/mL,甲状腺过氧化物酶抗体(A-TPO)>1 000 IU/mL。自身免疫性肝病IgG抗体(9项):抗线粒体M2抗体(+ + +),抗PML抗体(±),抗Sp100抗体(+ + +),抗线粒体M2-3E抗体(+ + +),抗gp210抗体(+ + +)。抗核抗体系列:抗nRNP/Sm(+ + +),颗粒型1∶ 3 200阳性,胞浆颗粒型1∶ 1 000阳性,着丝点抗体(CENPE)(+),抗线粒体M2抗体118.7 RU/mL,抗心磷脂IgM抗体71 MPL/mL,超敏C反应蛋白1.71 mg/L,IgG 25.86 g/L,IgM 7.01 g/L。血清蛋白电泳:γ-球蛋白32.17%;铁代谢:铁3.9 μmol/L,铁蛋白9.1 μg/L,总铁结合力150 μmol/L;肝功能:AST 90.4 U/L,ALT 104.1 U/L,GGT 321.6 U/L,ALP 505.5 U/L,胆碱酯酶6 950 U/L,白蛋白38 g/L,总胆红素26.8 μmol/L,直接胆红素7.6 μmo/L,间接胆红素19.2 μmo/L,抗β-2糖蛋白I抗体测定(IgG/A/M) 40 RU/mL。尿常规、粪便常规、凝血常规、肾功能、离子、叶酸、维生素B12、类风湿因子、抗链球菌溶血素O、抗中性粒细胞胞浆抗体(ANCA)确证和筛查,HBV定量均未见异常。
腹部彩超提示:肝实质回声粗密。甲状腺彩超提示:甲状腺实质弥漫性病变。全腹CT提示:(1)双侧附件略饱满,请结合生理期;双侧髂血管旁淋巴结显示,请结合临床;(2)心腔、大血管密度减低,考虑贫血,请结合临床。肺CT提示:(1)右肺中叶、左肺舌叶少许慢性炎症;(2)右肺中叶、左肺舌叶及下叶炎性小结节。骨髓涂片提示:(1)成熟红细胞轻度缗钱样排列,部分成熟红细胞中心淡染区扩大;(2)全片找到巨核细胞63个,分类50个,幼稚巨核15个,颗粒巨核35个,血小板散在少见;(3)铁染色示,外铁(-),内铁6%,未见环铁。诊断:巨核细胞成熟不良伴缺铁。骨髓活检:骨髓增生活跃,粒、红、巨核三系增生,未见异常淋巴细胞增多。血液病免疫分析初筛未见异常。网织血小板8.9%(正常参考值:12.50%±4.15%),PAIgG 11.8%(正常参考值:35.6%±7.4%), 血小板特异性自身抗体(流式法):GPⅨ阴性,GPⅢa阴性,GPⅠb阳性,GPⅡb阳性,GMPI40阳性。临床诊断:PBC、未分化结缔组织病、ITP、缺铁性贫血。
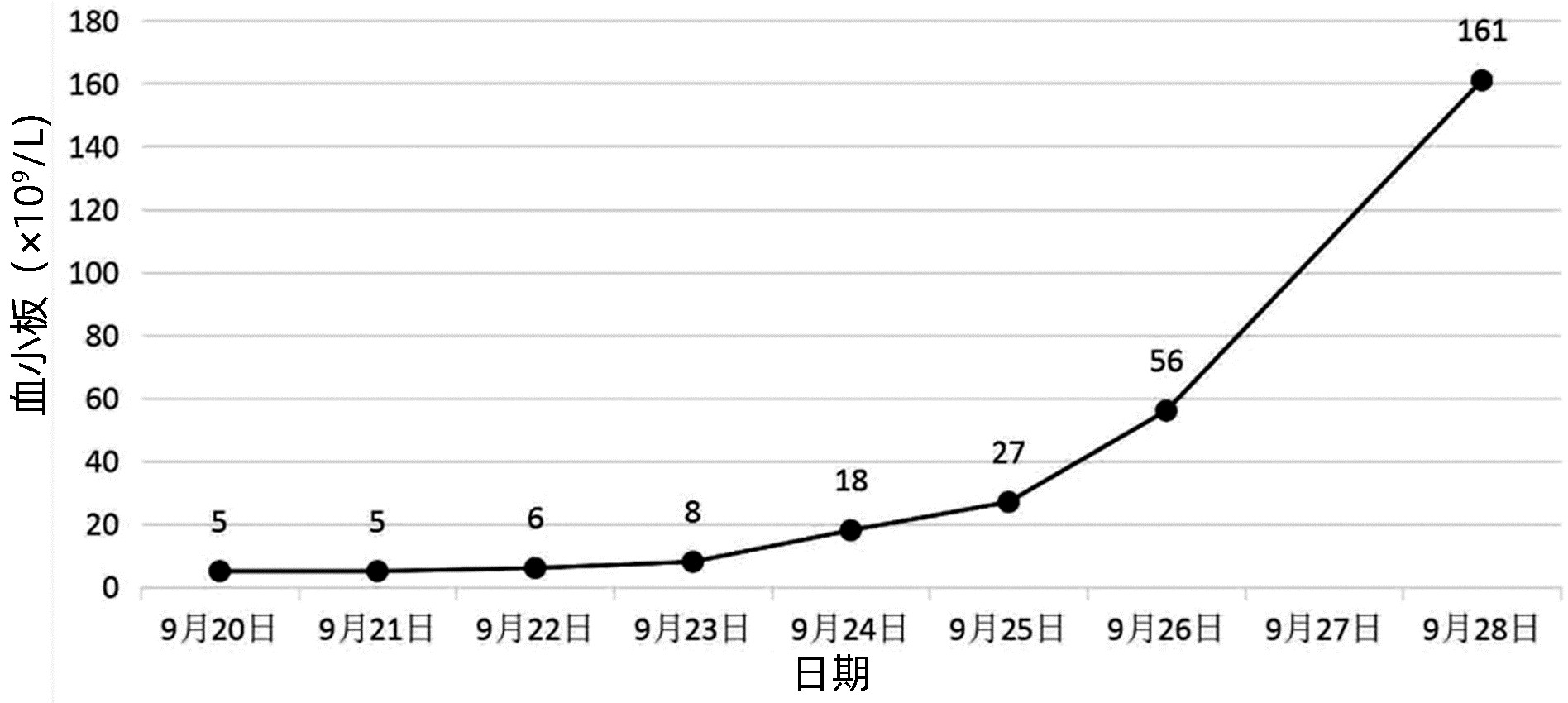
住院期间治疗:熊去氧胆酸胶囊、双环醇口服保肝、甲强龙60 mg/d静点(2022年9月20—28日);丙种球蛋白27.5 g,连续静点3天(2022年9月25—27日),艾斯奥美拉唑、聚普瑞锌保护胃黏膜、碳酸钙D3颗粒、骨化三醇预防骨质疏松,酚磺乙胺、白眉蛇毒血凝酶预防出血。经治疗后,患者未再出血,血小板恢复至正常水平(图 1),肝功能指标好转(图 2),出院后继续口服甲泼尼龙片60 mg/d,每周减量1次,羟氯喹0.2 g,2次/d口服,多糖铁胶囊1粒,2次/d口服,熊去氧胆酸胶囊500 mg,2次/d口服,同时辅以口服药物保护胃黏膜、补钙、预防感染等治疗;期间定期复查血常规、肝功能、肾功能、离子、血糖等,出院1个月后复查免疫五项、ANA系列,出院3个月后复查肺CT、骨密度,根据结果,风湿科门诊随诊调整口服药物。
2. 讨论
PBC是一种慢性自身免疫性肝内胆汁淤积性疾病,病因及发病机制尚未完全阐明,疾病进展的速度变化较大,通常病情进展缓慢[1]。PBC患者常受到至少一种额外的自身免疫性疾病的影响,包括干燥综合征、自身免疫性甲状腺疾病、类风湿关节炎和硬皮病等。一项国际多中心的回顾性研究[2]分析了1 554例PBC患者,440例PBC患者共诊断出了35种不同的肝外自身免疫疾病,有4例出现了ITP,264例合并了风湿系统疾病。在一项国内的单中心研究[3]中,共纳入分析了985例PBC患者,其中247例患者至少患有一种肝外自身免疫疾病,55例患者存在两种以上,220例合并了风湿系统疾病,干燥综合征是与PBC共存最常见的疾病,共140例,有4例患者合并ITP。PBC和肝外自身免疫疾病经常重叠,合并肝外自身免疫性疾病的患者多为女性,通常抗线粒体M2抗体、ANA的阳性率更高,该类患者确诊时的ALP、胆红素、AST水平和肝脏纤维化程度相对于不合并肝外自身免疫性疾病的PBC患者较低。PBC的典型症状为乏力和瘙痒,可严重影响生活质量,这些症状可能是相关肝外自身免疫性疾病的特征,而并非PBC的特征,PBC合并肝外自身免疫性疾病的早期诊断和疾病管理,对于改善患者生活质量具有重大意义[2, 4]。
PBC合并ITP比较少见,ITP是一种排除性诊断,单纯依靠骨髓穿刺、骨髓活检以及其他相关的实验室检查并不能作为ITP诊断的金标准,推荐患者可行血小板抗体的检测。2007年,国际ITP工作组将其诊断分为原发性与继发性两种,原发性是指患者无与血小板减少相关的疾病,是由自身免疫机制介导,以血小板减少和破坏增多为主要临床特征;而继发性是指由多种复杂因素或疾病共同导致的血小板减少,包括药物诱导,自身免疫性疾病(系统性红斑狼疮、干燥综合征等),淋巴细胞增殖性疾病(慢性淋巴细胞白血病和淋巴瘤)或病毒感染等。ITP的发病机制尚未完全阐明,免疫失调和自身抗体起着主要作用[2]。ITP患者血浆中存在多种自身抗体,在50%~70%的患者中,抗体识别一种或多种血小板表面糖蛋白(GP),包括GPⅡb Ⅲa、GPⅠb Ⅸ和GPⅠa Ⅱ。一般来说,脾功能亢进被怀疑是PBC血小板减少症的原因之一,但是血小板减少并不常见于没有脾功能亢进的早期PBC患者,然而一些PBC患者表现为血小板自身抗体(PAIgG)的升高,结合血小板下降的进展迅速,典型的骨髓象改变,血清PAIgG的升高,以及对类固醇治疗的快速反应,考虑免疫介导占主导地位。关于PBC血小板减少症的潜在机制,有研究表明血小板膜糖蛋白和线粒体蛋白M2具有相似的氨基酸序列,可能存在一个共同的抗体结合位点,出现交叉反应[5]。免疫遗传因素被认为会影响自身免疫性疾病的发生和发展,已经有研究发现,抗血小板表面糖蛋白自身抗体与人类白细胞抗原(human leukocyte antigen, HLA)等位基因之间有很强的相关性,PBC和ITP患者的2个HLA等位基因组合频率明显要高于普通人群。2个HLA等位基因组合可能是PBC和ITP发展的常见免疫遗传因素[6-9]。
免疫性疾病相关的ITP经典的一线治疗方法以糖皮质激素和静脉注射免疫球蛋白为主,其机制主要是抑制抗体介导的血小板破坏。对激素无效的患者可采取血小板生成素或血小板生成素受体的激动剂、利妥昔单抗、脾切除作为二线治疗。硫唑嘌呤、环孢素A、达那唑、长春新碱等免疫抑制剂也可以作为个体化的二线治疗[10]。
本例患者无明显的乏力、腹胀等肝病相关的症状,但肝功能提示GGT、ALP水平升高,排除了病毒、酒精、药物等常见肝损伤的原因后,筛查自身免疫性肝病抗体提示PBC特异性抗线粒体M2抗体、抗gp210抗体、抗Sp100抗体均为阳性,IgM升高,且影像未见明显的肝外和肝内胆管梗阻,综上,根据2021年中华医学会肝病学分会PBC指南[11],结果符合PBC诊断,患者目前有生化指标的异常,但没有明显的临床症状,处于无症状期,予以熊去氧胆酸和保肝治疗后,肝功能有明显改善。PBC患者常合并几种肝外的自身免疫性疾病,本例患者多项抗核抗体阳性,诊断考虑结缔组织病可能性大,但患者并没有相关的临床症状,仅予以患者羟氯喹口服预防疾病进展。患者本次主要因为“上肢散在瘀斑,鼻衄”入院,入院时血小板极低,患者无明显感染的表现,予以完善骨穿,血液病免疫分析初筛后,排除了恶性血液系统疾病(骨髓增生异常综合征、急慢性白血病)引起的血小板减少,且影像学检查未提示明确的肝脏形态改变,脾脏肿大,血常规提示单纯的血小板减少,白细胞和红细胞未见明显异常,故不考虑脾功能亢进导致的血小板破坏增多,患者抗核抗体系列阳性,血小板自身抗体阳性,自身免疫性肝病、结缔组织病诊断明确,患者免疫状态紊乱,予以激素联合人免疫球蛋白治疗后,血小板升至正常值,治疗有效,考虑患者ITP诊断明确。患者出院时血小板已恢复至正常值水平,截至目前,病情平稳。对于该类患者,长期口服激素治疗效果明显。通过该病例以期提高临床医生对该病的认识,及时诊断明确并针对性治疗,以免延误病情。
-
[1] KAPLAN MM, GERSHWIN ME. Primary biliary cirrhosis[J]. N Engl J Med, 2005, 353(12): 1261-1273. DOI: 10.1056/NEJMra043898. [2] EFE C, TORGUTALP M, HENRIKSSON I, et al. Extrahepatic autoimmune diseases in primary biliary cholangitis: prevalence and significance for clinical presentation and disease outcome[J]. J Gastroenterol Hepatol, 2021, 36(4): 936-942. DOI: 10.1111/jgh.15214. [3] CHEN S, LI MQ, DUAN WJ, et al. Concomitant extrahepatic autoimmune diseases do not compromise the long-term outcomes of primary biliary cholangitis[J]. Hepatobiliary Pancreat Dis Int, 2022, 21(6): 577-582. DOI: 10.1016/j.hbpd.2022.05.009. [4] WATT FE, JAMES OF, JONES DE. Patterns of autoimmunity in primary biliary cirrhosis patients and their families: a population-based cohort study[J]. QJM, 2004, 97(7): 397-406. DOI: 10.1093/qjmed/hch078. [5] PANZER S, PENNER E, NELSON PJ, et al. Identification of the platelet glycoprotein IIb/IIIa complex as a target antigen in primary biliary cirrhosis-associated autoimmune thrombocytopenia. Evidence that platelet-reactive autoantibodies can also bind to the mitochondrial antigen M2[J]. J Autoimmun, 1990, 3(4): 473-483. DOI: 10.1016/s0896-8411(05)80014-9. [6] KUWANA M, KABURAKI J, PANDEY JP, et al. HLA class Ⅱ alleles in Japanese patients with immune thrombocytopenic purpura. Associations with anti-platelet glycoprotein autoantibodies and responses to splenectomy[J]. Tissue Antigens, 2000, 56(4): 337-343. DOI: 10.1034/j.1399-0039.2000.560405.x. [7] TANAKA A, LEUNG P, GERSHWIN ME. The genetics of primary biliary cholangitis[J]. Curr Opin Gastroenterol, 2019, 35(2): 93-98. DOI: 10.1097/MOG.0000000000000507. [8] LI Y, LIU X, WANG Y, et al. Novel HLA-DRB1 alleles contribute risk for disease susceptibility in primary biliary cholangitis[J]. Dig Liver Dis, 2022, 54(2): 228-236. DOI: 10.1016/j.dld.2021.04.010. [9] MA WY, DENG ZH. Current status of immunogenetic studies on primary biliary cholangitis[J]. J Clin Hepatol, 2020, 36(4): 932-935. DOI: 10.3969/j.issn.1001-5256.2020.04.050.马伟煜, 邓志华. 原发性胆汁性胆管炎的免疫遗传研究现状[J]. 临床肝胆病杂志, 2020, 36(4): 932-935. DOI: 10.3969/j.issn.1001-5256.2020.04.050. [10] NEUNERT C, TERRELL DR, ARNOLD DM, et al. American Society of Hematology 2019 guidelines for immune thrombocytopenia[J]. Blood Adv. 2019. 3(23): 3829-3866. DOI: 10.1182/bloodadvances.2019000966. [11] Chinese Society of Hepatology, Chinese Medical Association. Guidelines on the diagnosis and management of primary biliary cholangitis (2021)[J]. J Clin Hepatol, 2022, 38(1): 35-41. DOI: 10.3760/cma.j.cn112138-20211112-00794. 期刊类型引用(0)
其他类型引用(7)
-

 下载:
下载:
 下载:
下载:

 本文二维码
本文二维码

计量
- 文章访问数: 498
- HTML全文浏览量: 113
- PDF下载量: 59
- 被引次数: 7

